El aprendizaje automático supervisado es una disciplina fundamental en la ciencia de datos y la IA. En este enfoque, trabajamos con un conjunto de datos en el que conocemos la variable objetivo que deseamos predecir o clasificar. Esta variable objetivo contiene la información que queremos explicar o entender, y su comportamiento se basa en el resto de las variables o características del conjunto de datos. El objetivo principal del ML supervisado es desarrollar modelos que puedan capturar patrones y relaciones entre las variables con el fin de realizar predicciones precisas y clasificar observaciones.
Uno de los aspectos más destacados del ML supervisado es su capacidad para resolver problemas de clasificación, donde se asignan observaciones a diferentes categorías o clases. Por ejemplo, podemos aplicar modelos de clasificación para predecir si un correo electrónico es spam o legítimo, si un paciente tiene una enfermedad específica o no, o si una transacción bancaria es fraudulenta o no.
Además de la clasificación, el ML supervisado también se utiliza para abordar problemas de regresión, donde la variable objetivo es una cantidad continua. Esto nos permite realizar predicciones numéricas, como predecir el precio de una casa en función de sus características, pronosticar la demanda de productos en función de variables de mercado, o estimar la duración de un proyecto en función de factores diversos.
En este tema, exploraremos diversos algoritmos de clasificación supervisada que permiten extraer patrones a partir de los datos y realizar predicciones precisas en una variedad de aplicaciones. Aprenderemos cómo entrenar modelos, evaluar su rendimiento y aplicarlos a situaciones del mundo real. El ML supervisado es una herramienta poderosa que puede proporcionar conocimientos valiosos y respuestas a preguntas importantes en una amplia gama de dominios.
7.1 Modelos lineales
Los modelos lineales representan uno de los pilares más robustos y extensamente empleados en el ámbito del aprendizaje supervisado en ML. Estos modelos constituyen la base de numerosas aplicaciones de predicción y clasificación, desempeñando un papel fundamental en la comprensión de los principios subyacentes de los algoritmos de ML.
La premisa fundamental de un modelo lineal es la suposición de que existe una relación lineal entre las variables de entrada (características) y la variable objetivo que se desea predecir. En otras palabras, se busca identificar una combinación lineal de las características ponderadas por coeficientes que se ajuste óptimamente a los datos observados. Los modelos lineales destacan por su alta interpretabilidad, lo que facilita un análisis claro sobre cómo cada característica influye en la variable objetivo.
En este capítulo exploraremos los modelos lineales, iniciando con los modelos lineales generalizados, entre los cuales el modelo de regresión lineal representa un caso específico. Además, examinaremos modelos lineales de clasificación, como la regresión logística, que se emplean para abordar problemas de clasificación.
7.1.1 Modelos lineales generalizados
Los Modelos Lineales Generalizados (GLM, de “Generalized Linear Models”) son una extensión poderosa de los modelos lineales tradicionales que amplían su aplicabilidad a una variedad de problemas más complejos en el campo del ML. Si necesitáis una referencia completa a este tipo de modelos: (McCullagh 2019).
Estos modelos son aplicables tanto a variables respuesta cuantitativas como cualitativas. De hecho, a diferencia de los modelos lineales simples, los GLM permiten manejar una amplia gama de situaciones, incluyendo variables de respuesta que no siguen una distribución normal, relaciones no lineales entre las características y la variable objetivo, y variables categóricas. Los GLM se basan en tres componentes clave:
la función de enlace,
la distribución de probabilidad,
la estructura lineal.
Llamamos \(y\) a la variable respuesta. Además, se define un predictor lineal con la combinación lineal ponderada de las \(p\) variables explicativas \(x_1, x_2, ..., x_p\):
La función de enlace conecta la media de la variable respuesta con la combinación lineal de las características (el predictor lineal), lo que permite modelar relaciones no lineales y capturar la variabilidad de los datos. La elección de la función de enlace depende del tipo de problema que se esté abordando y puede incluir funciones como la logística para problemas de clasificación o la identidad para problemas de regresión.
La estructura lineal implica que las características se ponderan por coeficientes, similar a los modelos lineales tradicionales, pero con la capacidad de ajustar relaciones no lineales y manejar múltiples predictores, incluyendo variables categóricas.
PrecauciónRepaso
Quizás es un buen momento para repasar los modelos lineales (regresión) que hayas visto en otras asignaturas del grado.
Definimos una función de enlace que relaciona el valor esperado de la variable respuesta dadas las variables explicativas \(E(y|x)=\mu(x)\), con el predictor lineal:
\[g(E(y|x))=\eta(x)\]
La distribución de probabilidad (componente aleatoria) describe cómo se distribuyen los datos alrededor de la media y puede adaptarse a diferentes tipos de datos, como datos binarios, Poisson, binomiales o exponenciales, entre otros. Esto brinda flexibilidad para modelar una variedad de escenarios de datos. Por ejemplo, en el caso de la conocida regresión lineal se asume que la variable respuesta sigue una distribución Normal. Mediante la componente aleatoria determinamos cómo añadir el ruido o error aleatorio a la predicción que se obtiene de la función de enlace. La tabla siguiente, adaptada de (Agresti 2015), resume los GLM:
Modelo
Componente aleatoria
Función de enlace
Tipo de variables explicativas
Regresión Lineal
Normal
Identidad
Cuantitativas
ANOVA
Normal
Identidad
Cuantitativas
ANCOVA
Normal
Identidad
Cuantitativas y Cualitativas
Regresión Logística
Binomial
Logit
Cuantitativas y Cualitativas
LogLinear
Poisson
Log
Cualitativas
Regresión de Poisson
Poisson
Log
Cuantitativas y Cualitativas
ImportantePara recordar
Los GLM no asumen una relación lineal entre la variable respuesta y las variables explicativas (o independientes), pero sí suponen una relación lineal entre la variable respuesta transformada en términos de la función de enlace y las variables explicativas.
7.1.2 Regresión Logística
El modelo de Regresión Logística es uno de los modelos lineales generalizados más empleado en la práctica por la facilidad en la interpretación de sus resultados. Es un modelo adecuado para modelar la relación entre una variable respuesta binaria (\(0\) o \(1\)) y un conjunto de variables explicativas cuantitativas y/o cualitativas.
NotaMulticlase
Existe una versión de la Regresión Logística adaptada al caso de una variable respuesta con más de dos categorías, que recibe el nombre de Regresión Logística Politómica o Multinomial.
Asumiremos que la variables explicativas son independientes entre sí y que la probabilidad de \(1\) (a veces llamada probabilidad de éxito) solo depende de los valores de dichas variables. Si las variables explicativas no fueran independientes, podríamos añadir interacciones al modelo.
Tratamos de modelar la probabilidad condicionada, \(\mu(x)\), de que la variable respuesta sea un \(1\) dados los valores de las variables explicativas. En este caso, la componente de la variables respuesta se corresponde con una distribución binomial, \(Binomial(n,\pi)\), donde \(\pi=\mu(x)\) es la probabilidad de éxito. Así,
que modela la log odds de probabilidad de \(1\) como función de las variables explicativas. Esto es, se modela el logaritmo de la probabilidad de \(1\) dadas las variables explicativas, frente a la probabilidad de \(0\) dadas las variables explicativas.
Se emplean métodos de Máxima Verosimilitud para la estimación de los parámetros \(\beta_i, i=0,...,p\). Es posible realizar contrastes de hipótesis sobre dichos parámetros, tratando de eliminar del modelo las variables no significativas. Es decir, se contrasta si en los datos hay suficiente información contraria a la siguiente hipótesis nula, como para rechazarla: \[H_o:\beta_i=0\] La interpretación de los coeficientes se realiza mediante una razón de ventajas (“odds ratio” en inglés), tomando la exponencial de los estimadores de los parámetros del modelo.
Con los parámetros estimados, y sus desviaciones típicas, podremos hacer selección de variables, quedándonos con las más significativas (aquellas con un \(p-valor\) por debajo de un umbral).
Vamos a trabajar con el ejemplo de los bancos que se estudió en el Capítulo 3. Como primera aproximación a la regresión logística, aplicamos un modelo con una única variable explicativa. En este caso elegimos la variable housing:
#Lectura de datoslibrary(tidyverse)bank =read.csv('https://raw.githubusercontent.com/rafiag/DTI2020/main/data/bank.csv')dim(bank)
[1] 11162 17
bank=as.tibble(bank)# Parciticionamos los datosset.seed(2138)n=dim(bank)[1]indices=seq(1:n)indices.train=sample(indices,size=n*.5,replace=FALSE)indices.test=sample(indices[-indices.train],size=n*.25,replace=FALSE)indices.valid=indices[-c(indices.train,indices.test)]bank.train=bank[indices.train,]bank.test=bank[indices.test,]bank.valid=bank[indices.valid,]# Estudiamos la influencia de housing en deposittabla1=xtabs(~housing+deposit,data=bank.train)tabla1
deposit
housing no yes
no 1246 1688
yes 1662 985
chisq.test(tabla1)
Pearson's Chi-squared test with Yates' continuity correction
data: tabla1
X-squared = 229.44, df = 1, p-value < 2.2e-16
Podemos observar que existe una relación relevante entre ambas variables. El \(p-valor\) es, claramente, inferior al valor de referencia \(0.05\). Empleamos la regresión logística para cuantificar la relación:
factor.deposit <-factor(bank.train$deposit)logit1 <-glm(factor.deposit ~ housing, data = bank.train, family ="binomial")summary(logit1)
Call:
glm(formula = factor.deposit ~ housing, family = "binomial",
data = bank.train)
Coefficients:
Estimate Std. Error z value Pr(>|z|)
(Intercept) 0.30361 0.03735 8.129 4.34e-16 ***
housingyes -0.82674 0.05488 -15.064 < 2e-16 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
(Dispersion parameter for binomial family taken to be 1)
Null deviance: 7727 on 5580 degrees of freedom
Residual deviance: 7495 on 5579 degrees of freedom
AIC: 7499
Number of Fisher Scoring iterations: 4
¿Qué vemos en la salida? En primer lugar, los 3 asteriscos (” *** “) en la fila correspondiente a la variable housing indican su alta significatividad estadística. En otras palabras, es una variable muy relacionada con la respuesta (como ya habíamos averiguado). El valor del parámetro \(\beta_1\) es de \(-0.82674\) con una error estándar de \(0.05488\). ¿Qué significa? Es complicado interpretar valores negativos en los coeficientes. Para una mayor comprensión vamos a darle la vuelta. Es decir, el valor \(-0.82674\) está asociado a”housing=yes”, de modo que el valor \(0.82674\) está asociado a “housing=no”. La interpretación se realiza en base al exponente de dicho valor tal y como sigue: \[exp(.82674)=2.29\] significa que el “riesgo” de contratar un depósito es \(2.29\) veces mayor en aquellos que no tienen casa, en relación con el mismo riesgo para aquellos que sí tienen casa en propiedad. Si hubíeramos mantenido la interpretación original (con el valor del coeficiente negativo), el resultado sería: “el riesgo de contratar un depósito es \(exp(-0.82674)=0.44\) veces menor en aquellos propietarios de su propia casa, respecto a otros clientes que no tienen casa. Como puede verse es mucho más sencillo interpretar coeficientes positivos (odds ratio mayor que \(1\)) que coeficientes negativos.
En base al valor del parámetro y a su error, podemos obtener intervalos de confianza:
Y por tanto, el riesgo de contratar un depósito es \(2.29 \in [2.05,2.54]\) veces mayor en aquellos que no tienen casa, en relación con el mismo riesgo para aquellos que sí tienen casa en propiedad.
Vamos ahora a trabajar en el caso multivariante. Como primer paso estudiemos, una a una las variables que se van a incluir en el modelo. A modo de ejemplo, consideramos las siguientes variables:
housing
marital
education
impago
saldo
# para que la categoría de referencia sea "married"bank.train$marital=relevel(as.factor(bank.train$marital),ref=2)logit2 <-glm(factor.deposit ~ marital, data = bank.train, family ="binomial")summary(logit2)
Call:
glm(formula = factor.deposit ~ marital, family = "binomial",
data = bank.train)
Coefficients:
Estimate Std. Error z value Pr(>|z|)
(Intercept) -0.24428 0.03575 -6.832 8.35e-12 ***
maritaldivorced 0.19139 0.08662 2.209 0.0271 *
maritalsingle 0.43555 0.05974 7.290 3.09e-13 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
(Dispersion parameter for binomial family taken to be 1)
Null deviance: 7727.0 on 5580 degrees of freedom
Residual deviance: 7673.4 on 5578 degrees of freedom
AIC: 7679.4
Number of Fisher Scoring iterations: 3
La variable marital es estadísticamente significativa. Los clientes solteros tienen un riesgo del orden de \(1.5\) veces mayor de deposit=yes respecto a los clientes casados. Los clientes divorciados tienen un riesgo del orden de \(1.2\) veces mayor.
logit2 <-glm(factor.deposit ~ education, data = bank.train, family ="binomial")summary(logit2)
Call:
glm(formula = factor.deposit ~ education, family = "binomial",
data = bank.train)
Coefficients:
Estimate Std. Error z value Pr(>|z|)
(Intercept) -0.42876 0.07457 -5.750 8.92e-09 ***
educationsecondary 0.25901 0.08385 3.089 0.002008 **
educationtertiary 0.59332 0.08820 6.727 1.73e-11 ***
educationunknown 0.48005 0.14220 3.376 0.000736 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
(Dispersion parameter for binomial family taken to be 1)
Null deviance: 7727 on 5580 degrees of freedom
Residual deviance: 7671 on 5577 degrees of freedom
AIC: 7679
Number of Fisher Scoring iterations: 4
La variable education es estadísticamente significativa. A medida que la educación aumenta, los clientes tienen un riesgo mayor de deposit=yes (\(1\) categoría de referencia, \(1.3\) educación secundaria, \(1.8\) educación terciaria). Los clientes con nivel de educación desconocido también tienen un riesgo positivo. Aquí la interpretación es más complicada al desconocer el nivel real de educación de estos clientes.
logit2 <-glm(factor.deposit ~ default, data = bank.train, family ="binomial")summary(logit2)
Call:
glm(formula = factor.deposit ~ default, family = "binomial",
data = bank.train)
Coefficients:
Estimate Std. Error z value Pr(>|z|)
(Intercept) -0.07800 0.02701 -2.887 0.00388 **
defaultyes -0.39200 0.21713 -1.805 0.07102 .
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
(Dispersion parameter for binomial family taken to be 1)
Null deviance: 7727.0 on 5580 degrees of freedom
Residual deviance: 7723.7 on 5579 degrees of freedom
AIC: 7727.7
Number of Fisher Scoring iterations: 3
La variable default no es estadísticamente significativa. Su \(p-valor\) es mayor que \(0.05\). Sin embargo, está cerca de dicho valor de referencia. Eliminar esta variable del modelo en una etapa tan temprana puede ser un error.
logit2 <-glm(factor.deposit ~ balance, data = bank.train, family ="binomial")summary(logit2)
Call:
glm(formula = factor.deposit ~ balance, family = "binomial",
data = bank.train)
Coefficients:
Estimate Std. Error z value Pr(>|z|)
(Intercept) -1.667e-01 3.036e-02 -5.489 4.04e-08 ***
balance 5.621e-05 1.004e-05 5.600 2.15e-08 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
(Dispersion parameter for binomial family taken to be 1)
Null deviance: 7727.0 on 5580 degrees of freedom
Residual deviance: 7689.1 on 5579 degrees of freedom
AIC: 7693.1
Number of Fisher Scoring iterations: 3
La variable saldo es claramente significativa. Un incremento de una unidad en el saldo está asociado a un incremento de \(1.00006\) en el riesgo de depóstivo. En variables continuas es muy habitual que un incremento tan bajo (\(1\) dolar esté asociado a incrementos tan pequeños en el riesgo). Para mejorar la interpretación se suele multiplicar esta cantidad como sigue:
exp(logit2$coefficients[2]*1000)
balance
1.057816
De este modo, la interpretación sería: los clientes con \(1000\) dólares más en el salto incrementan su riesgo de depósito en \(1.06\) respecto a los que tienen \(1000\) dólares menos.
PrecauciónMétodos Wrapper de selección de características
En el Capítulo 4 vimos algunas técnicas de reducción de la dimensión basadas en la selección de características de acuerdo con la calidad de las mismas. Esta calidad se evalua empleando algoritmos de ML.
A continuación, ajustamos un modelo de regresión logística con todas las características significativas.
logit2 <-glm(factor.deposit ~ housing+marital+education+default+ balance, data = bank.train, family ="binomial")summary(logit2)
Call:
glm(formula = factor.deposit ~ housing + marital + education +
default + balance, family = "binomial", data = bank.train)
Coefficients:
Estimate Std. Error z value Pr(>|z|)
(Intercept) -1.982e-01 8.389e-02 -2.362 0.01815 *
housingyes -7.770e-01 5.585e-02 -13.912 < 2e-16 ***
maritaldivorced 1.776e-01 8.901e-02 1.995 0.04604 *
maritalsingle 3.728e-01 6.211e-02 6.002 1.95e-09 ***
educationsecondary 2.475e-01 8.666e-02 2.856 0.00429 **
educationtertiary 4.323e-01 9.166e-02 4.717 2.40e-06 ***
educationunknown 3.178e-01 1.464e-01 2.170 0.03000 *
defaultyes -2.920e-01 2.222e-01 -1.314 0.18872
balance 4.495e-05 1.007e-05 4.462 8.12e-06 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
(Dispersion parameter for binomial family taken to be 1)
Null deviance: 7727.0 on 5580 degrees of freedom
Residual deviance: 7397.1 on 5572 degrees of freedom
AIC: 7415.1
Number of Fisher Scoring iterations: 4
Podemos ver como la variable default pierde significatividad estadística. Esto es normal. Al tener en consideración el efecto de otras variables, aquellas que estaban al límite de la significatividad pueden ser eliminadas. Entrenamos el modelo sin esa variable.
logit2 <-glm(factor.deposit ~ housing+marital+education+ balance, data = bank.train, family ="binomial")summary(logit2)
Call:
glm(formula = factor.deposit ~ housing + marital + education +
balance, family = "binomial", data = bank.train)
Coefficients:
Estimate Std. Error z value Pr(>|z|)
(Intercept) -2.042e-01 8.377e-02 -2.438 0.01479 *
housingyes -7.772e-01 5.584e-02 -13.917 < 2e-16 ***
maritaldivorced 1.751e-01 8.898e-02 1.968 0.04908 *
maritalsingle 3.735e-01 6.209e-02 6.014 1.80e-09 ***
educationsecondary 2.479e-01 8.667e-02 2.861 0.00423 **
educationtertiary 4.328e-01 9.167e-02 4.721 2.35e-06 ***
educationunknown 3.151e-01 1.464e-01 2.152 0.03137 *
balance 4.582e-05 1.009e-05 4.543 5.54e-06 ***
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
(Dispersion parameter for binomial family taken to be 1)
Null deviance: 7727.0 on 5580 degrees of freedom
Residual deviance: 7398.8 on 5573 degrees of freedom
AIC: 7414.8
Number of Fisher Scoring iterations: 4
Todas las variables del modelo son estadísticamente significativas. Si las variables fueran independientes, los coeficientes de la regresión múltiple coincidirían al 100% con los coeficientes de las regresiones simples. Por ejemplo, el coeficiente asociado a la variable housing era \(2.29\) y ahora es \(2.18\), muy similar. Podemos intuir la existencia de una pequeña correlación entre las variables del modelo.
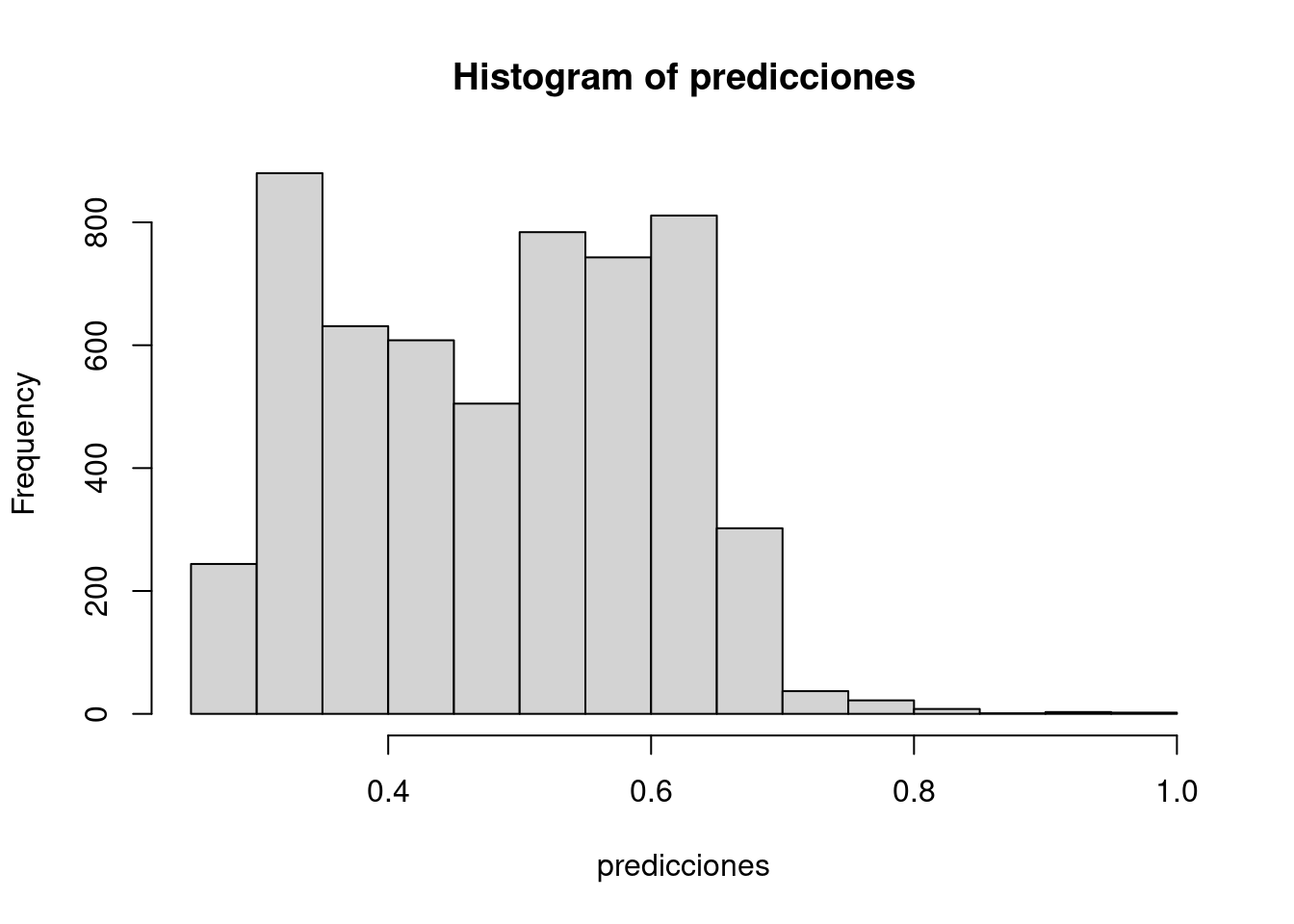
7.1.3 Probabilidad de clase
Podemos calcular la probabilidad que ofrece el modelo para cada una de las observaciones en la muestra de entrenamiento.
Confusion Matrix and Statistics
Reference
Prediction no yes
no 1790 1078
yes 1118 1595
Accuracy : 0.6065
95% CI : (0.5936, 0.6194)
No Information Rate : 0.5211
P-Value [Acc > NIR] : <2e-16
Kappa : 0.2121
Mcnemar's Test P-Value : 0.4053
Sensitivity : 0.5967
Specificity : 0.6155
Pos Pred Value : 0.5879
Neg Pred Value : 0.6241
Prevalence : 0.4789
Detection Rate : 0.2858
Detection Prevalence : 0.4861
Balanced Accuracy : 0.6061
'Positive' Class : yes
En la salida tenemos algunas de las medidas de rendimiento estudiadas en el Capítulo 6. Sin embargo, no aparece el \(F_1-score\), media armónica de la Precisión y la Recuperación. En cualquier caso, es fácil de programar:
Este es el funcionamiento del modelo en entrenamiento, pero ¿cuál es su funcionamiento en la partición de prueba? Recuerda, si el valor es mucho menor entonces estamos sobreajustando el modelo de entrenamiento.
Vemos que el valor es muy similar al obtenido en entrenamiento. No estamos sobreajustando el modelo.
PrecauciónTarea
Dejamos como labor del estudiante la tarea de mejorar la capacidad predictiva del modelo. Puedes emplear algunas de las variables que no han sido consideradas y que puedes consultar en el Capítulo 3.
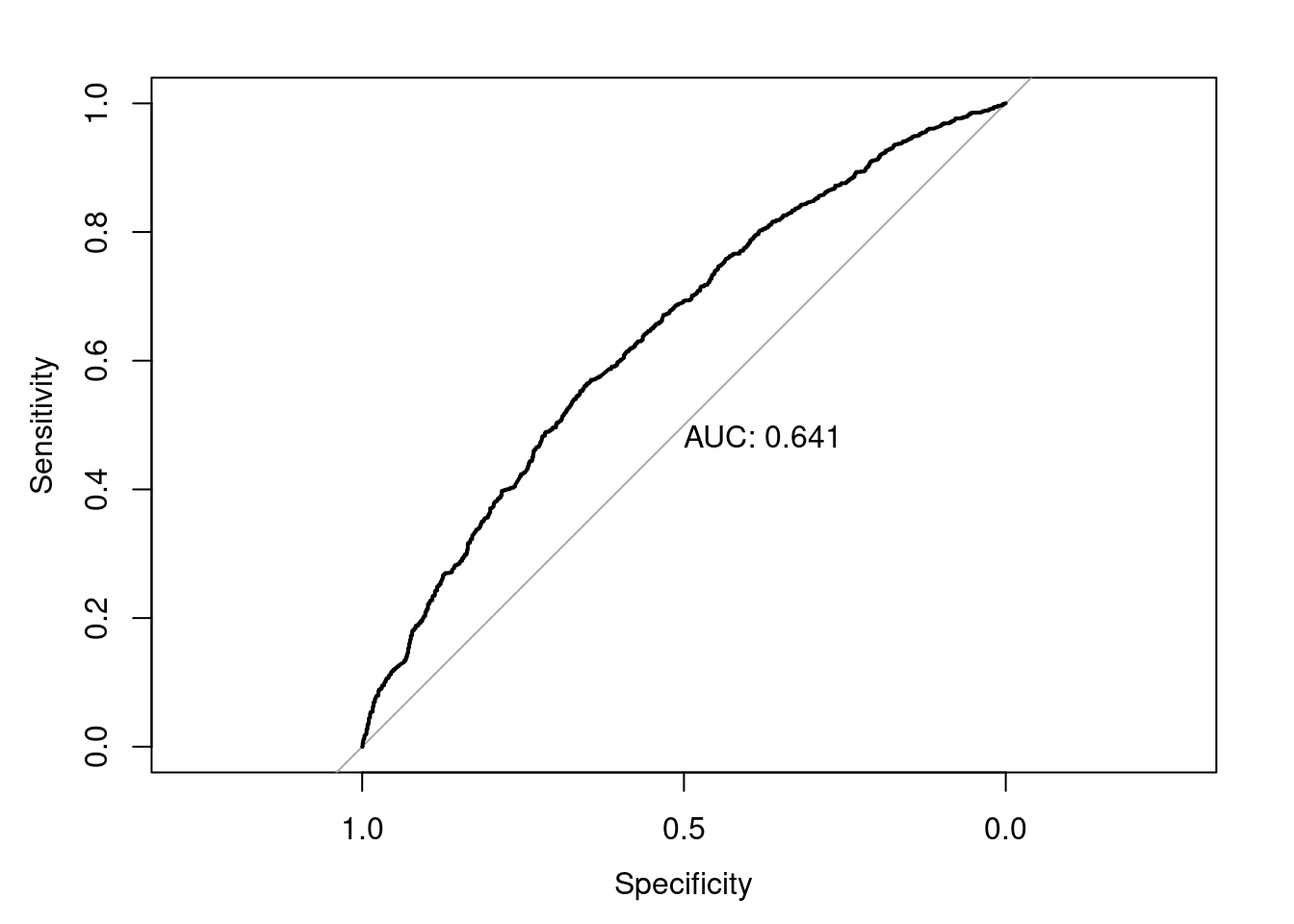
Podemos calcular la curva ROC presentada en el Capítulo 6.
Y ahora, podemos plantear si este modelo es adecuado. Es decir ¿estamos satisfechos con el rendimiento del modelo? ¿Es suficientemente bueno?
7.1.4 Análisis Discriminante Lineal
El análisis discriminante lineal (Linear Discriminant Analysis, LDA)(Hastie et al. 2009), (Murphy 2012) es una técnica estadística de clasificación supervisada utilizada para encontrar una combinación lineal de variables que discrimine dos o más grupos. Se basa en la idea de modelar la distribución de cada clase y aplicar el criterio de Bayes para asignar nuevas observacones a una de ellas.Se trata de un algoritmo de clasificación, es decir, la variable objetivo \(Y\) es categórica: \(Y \in \{1,\dots,C\}\).
Supongamos que los datos de cada clase \(Y=c\) siguen una distribución Normal multivariante:
\(\mathbf{x} \in \mathbb{R}^{p}\) es una observación
\(\mu_c\) es la media de la clase \(c\)
\(\Sigma_c\) es la matriz de covarianzas de la clase \(c\)
LDA surge cuando asumimos que las clases tienen una matriz de covarianzas común, es decir, \(\Sigma_c=\Sigma\), \(\forall c\).
El LDA se basa en el Teorema de Bayes. Bajo este teorema, asignaremos una observación \(\mathbf{x}\) a la clase que maximiza la probabilidad a posteriori \(P(Y=c|\mathbf{x})\) que, por el teorema de Bayes es
\[ P(Y=c|\mathbf{X}=\mathbf{x})=\frac{P(\mathbf{X}=\mathbf{x}| Y =c)P(Y=c)}{P(\mathbf{x})}\]
Así, si queremos comparar 2 clases, la \(0\) y la \(1\), bastará con analizar el siguiente log-ratio: \[log\left( \frac{P(Y=1|\mathbf{X}=\mathbf{x})}{P(Y=0|\mathbf{X}=\mathbf{x})} \right) = log\left( \frac{P(Y=1|\mathbf{x})}{P(Y=0|\mathbf{x})} \right) + log\left( \frac{P(Y=1)}{P(Y=0)} \right) =\]
Obtenemos una ecuación que es lineal en \(\mathbf{x}\). Puede verse más fácilmente llamando: \(\mathbf{w}=\Sigma^{-1}(\mu_1-\mu_0)\) y \(w_0 = log\left( \frac{P(1)}{P(0)} \right) - \frac{1}{2}(\mu_1 + \mu_0)^{t}\Sigma^{-1}(\mu_1-\mu_0)\).
Esta ecuación define la frontera de decisión lineal donde la probabilidad de las clases es igual cuando \(\mathbf{x}^{t}\mathbf{w}+w_0 =0\). La frontera de decisión entre las clases \(0\) y \(1\) es una ecuación lineal, un hiperplano en dimensión \(p\).
¿Cómo se lleva entonces a cabo la clasificación? Como la frontera es un hiperplano separador, se comparará a qué población está más cercana cada observación. Esto es, para clasificar una observación como perteneciente a la clase \(1\), tendrá que ocurrir que
Con esto, de forma general, vemos que las funciones discriminantes lineales son \[\delta_{c}(\mathbf{x})=\mathbf{x}^{t} \Sigma^{-1}\mu_{c} -\frac{1}{2}\mu_{c}^{t}\Sigma^{-1}\mu_{c} + \log(P(Y=c))\]
Esta es la función que se maximiza para asignar la observación \(\mathbf{x}\) a la clase de mayor probabilidad. Esto es, la regla de decisión es \(argmax_{c} \delta_{c}(\mathbf{x})\), que significa que se asignará \(\mathbf{x}\) a la clase \(c\) que tenga el mayor valor de \(\delta_{c}(\mathbf{x})\). La ecuación de la frontera de decisión vista antes es justamente la que se obtiene cuando \(\delta_{0}(\mathbf{x}) =\delta_{1}(\mathbf{x})\).
En la práctica, se desconocen los parámetros de la distribución Normal. Se estiman con los datos de entrenamiento:
\(\widehat{P(c)} = n_c/n\), siendo \(n_c\) el tamaño de la clase \(c\) y \(n\) el total
\(\widehat{\mu}_c\) media muestral de los elementos de la clase \(c\)
\(\widehat{\Sigma}=\frac{1}{n-C}\sum_{c=1}^{C}\sum_{y_i = c}(\mathbf{x}_i - \widehat{\mu}_c)(\mathbf{x}_i - \widehat{\mu}_c)^{t}\). Se utilizan los datos de todas las clases para calcular la matriz de covarianzas: suma de las matrices de covarianza de cada clase, ponderadas según su tamaño muestral (MLE). Se resta el número total clases \(C\) porque es el número de medias \(\widehat{\mu}_c\) estimado.
NotaVisión geométrica del LDA
El Análisis Discriminante Lineal (LDA) sigue una filosofía similar al Análisis de Componentes Principales (PCA), pero con un objetivo distinto: en lugar de buscar la máxima variabilidad de los datos sin considerar etiquetas, LDA se centra en maximizar la separabilidad entre clases. Es decir, busca encontrar una proyección de los datos en una nueva dimensión en la que las clases queden lo más separadas posible.
Una forma intuitiva de ver el LDA es como una proyección geométrica de los datos en una recta, donde la asignación de clase se realiza según la proximidad a las medias proyectadas de cada clase. Matemáticamente, se busca un vector de proyección \(\mathbf{w}\) tal que los datos proyectados \(\mathbf{z}=\mathbf{w}^t\mathbf{x}\) de distintas clases queden lo más separados posibles. \(\mathbf{z}_i\) representa la coordenada del punto \(\mathbf{x}_i\) en la nueva dimensión reducida. ¿Te recuerda a PCA? En efecto, en ambos casos se realiza una transformación lineal de los datos. Sin embargo, mientras que en PCA se maximizan las varianzas de los datos proyectados, en LDA se maximiza la separación entre clases y se minimiza la dispersión dentro de cada clase.
Para encontrar el nuevo eje de proyección, LDA busca maximizar la distancia entre las medias de las clases y minimizar la dispersión dentro de cada una de ellas. En el caso binario, definimos las medias de cada clase como: \[\mu_{0} = \frac{1}{n_{0}} \sum_{i:Y_{i}=0} \mathbf{x}_i\] y \[\mu_{1} = \frac{1}{n_{1}} \sum_{i:Y_{i}=1} \mathbf{x}_i\] denotando \(i:Y_{i}=0\) aquellas observaciones \(i\) que son de la clase \(0\), es decir, \(Y_i=0\) y siendo \(n_0\) y \(n_1\) los tamaños de las clases \(0\) y \(1\), respectivamente.
Si proyectamos estas medias sobre el nuevo eje definido por \(\mathbf{w}\), obtenemos \(m_c = \mathbf{w}^{t}\mu_{c}\).
Para evaluar la dispersión de los datos en la nueva dimensión, definimos la varianza de los puntos proyectados en cada clase como: \[S_{c}^{2}=\sum_{i:Y_i=c}(z_i-m_c)^{2}.\]
Dado que queremos que las medias proyectadas estén lo más separadas posible y que la dispersión dentro de cada clase sea mínima, definimos la función objetivo como: \[J(\mathbf{w})=\frac{(m_1-m_0)^{2}}{S_{0}^{2} + S_{1}^{2}}.\] Esta expresión se puede reformular en términos matriciales: \[J(\mathbf{w})=\frac{\mathbf{w}^{t}S_{B}\mathbf{w}}{\mathbf{w}^{t}S_{W}\mathbf{w} },\] donde:
\(S_{B}=(\mu_1 - \mu_0)(\mu_1 - \mu_0)^{t}\) es la matriz de dispersión entre las clases (between-class scatter)
y \(S_{W}=\sum_{i:Y_i=0}(\mathbf{x}_i - \mu_0)(\mathbf{x}_i - \mu_0)^{t} + \sum_{i:Y_i=1}(\mathbf{x}_i - \mu_1)(\mathbf{x}_i - \mu_1)^{t}\) es la matriz de dispersión dentro de cada clase (within-class).
Esto es así puesto que \[\mathbf{w}^{t}S_{B}\mathbf{w} = \mathbf{w}^{t}(\mu_1 - \mu_0)(\mu_1 - \mu_0)^{t}\mathbf{w}=(m_1 - m_0)^{2}\] y, por otro lado, \[\mathbf{w}^{t}S_{W}\mathbf{w} = \sum_{i:Y_i = 0}\mathbf{w}^{t}(\mathbf{x}_i - \mu_0)(\mathbf{x}_i - \mu_0)^{t}\mathbf{w} + \sum_{i:Y_i = 1}\mathbf{w}^{t}(\mathbf{x}_i - \mu_1)(\mathbf{x}_i - \mu_1)^{t}\mathbf{w}=\]\[=\sum_{i:Y_i = 0} (\mathbf{z}_i-m_0)^{2}+\sum_{i:Y_i = 1} (\mathbf{z}_i-m_1)^{2}\]
Para maximizar \(J(\mathbf{w})\), derivamos con respecto \(\mathbf{w}\) y llegamos a la ecuación característica \(S_{B}\mathbf{w}=\lambda S_{W}\mathbf{w}\). Si \(S_{W}\) es invertible, \(S_{W}^{-1}S_{B}\mathbf{w}=\lambda \mathbf{w}\). Esto recuerda al problema de autovalores y autovectores de PCA. Sin embargo, en este caso, como \(S_{B}\mathbf{w}=(\mu_1 - \mu_0)(\mu_1 - \mu_0)^{t}\mathbf{w}=(\mu_1 - \mu_0)(m_1 - m_0)\). Podemos sustituir en la ecuación característica y llegamos a que \[\lambda \mathbf{w}=S_{W}^{-1}(\mu_1 - \mu_0)(m_1 - m_0).\] Es decir, \[\mathbf{w} \propto S_{W}^{-1}(\mu_1 - \mu_0).\] En este punto, como el objetivo es encontrar la dirección de la proyección, tomamos \(\mathbf{w}=S_{W}^{-1}(\mu_1 - \mu_0)\), que es lo que obtuvimos antes.
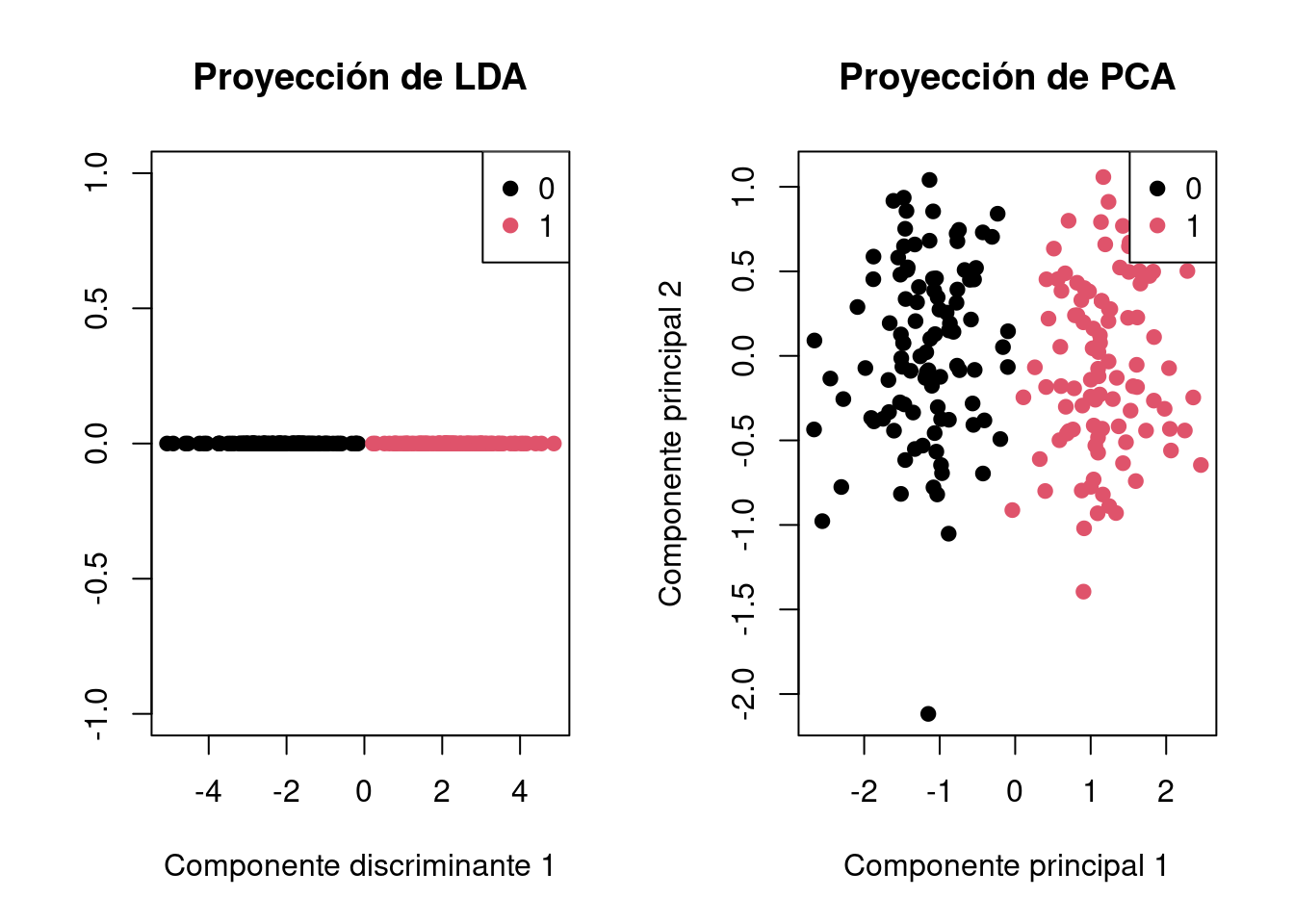
Veamos ahora un ejemplo para compara LDA y PCA.
# Libreríaslibrary(MASS)
Attaching package: 'MASS'
The following object is masked from 'package:dplyr':
select
# Semilla para reproducibilidadset.seed(42)# Conjunto de datos artificialn <-100# número de observaciones por clasep <-2# número de variablesclass_0 <-matrix(rnorm(n * p, mean =0), ncol = p)class_1 <-matrix(rnorm(n * p, mean =3), ncol = p)data <-rbind(class_0, class_1)labels <-factor(c(rep(0, n), rep(1, n)))df <-data.frame(x1 = data[, 1], x2 = data[, 2], class = labels)# Visualización de los datos originalesplot(df$x1, df$x2, col = df$class, pch =19, xlab ="Variable 1", ylab ="Variable 2", main ="Datos Originales")legend("topright", legend =levels(df$class), col =1:length(levels(df$class)), pch =19)
# LDAlda_model <-lda(class ~ x1 + x2, data = df)# Puntuaciones proyectadas en el espacio discriminante de LDAlda_predictions <-predict(lda_model)# PCApca_model <-prcomp(df[, -3], center =TRUE, scale. =TRUE)# Puntuaciones proyectadas en el espacio de PCApca_predictions <- pca_model$x# Graficar LDA y PCApar(mfrow =c(1, 2))# Gráfico de LDAplot(lda_predictions$x[, 1], rep(0, length(lda_predictions$x[, 1])), col = df$class, xlab ="Componente discriminante 1", ylab ="", main ="Proyección de LDA", pch =19)legend("topright", legend =levels(df$class), col =1:length(levels(df$class)), pch =19)# Gráfico de PCAplot(pca_predictions[, 1], pca_predictions[, 2], col = df$class,xlab ="Componente principal 1", ylab ="Componente principal 2", main ="Proyección de PCA", pch =19)legend("topright", legend =levels(df$class), col =1:length(levels(df$class)), pch =19)
par(mfrow =c(1, 1))
NotaVentajas
Simple y rápido
Eficiente cuando se cumple las hipótesis
Clasificación de observaciones en grupos determinados. LDA se utiliza comúnmente para clasificar observaciones en grupos predeterminados, lo que facilita la interpretación y la toma de decisiones.
Combinación de información para la frontera de decisión. Al considerar la información combinada de varias variables, LDA puede capturar patrones que podrían no ser evidentes al analizar cada variable por separado
NotaDesventajas
Asume normalidad y homocedasticidad. LDA puede ser sensible a la falta de normalidad en las variables y a la diferente variabilidad en las mismas.
Sensible a outliers. LDA puede ser sensible a la presencia de valores atípicos en los datos, lo que puede afectar negativamente la calidad del modelo.
Es un clasificador lineal.Como su nombre indica, LDA es lineal y puede no funcionar bien en situaciones donde la relación entre las variables predictoras y la variable dependiente es no lineal.
Requiere cierto tamaño de muestra. Para obtener resultados confiables, LDA requiere un tamaño de muestra adecuado en cada grupo, y puede no funcionar bien con tamaños de muestra desequilibrados
7.2 k-Vecinos
El método de los \(k\) vecinos más cercanos (\(k-NN\)), abreviatura de “k nearest neighbors” en inglés, se cuenta entre los enfoques más simples y ampliamente utilizados en el campo del ML. Este método se basa en la noción de similitud (o distancia) entre observaciones.
ImportantePara recordar
La principal asunción del modelo de los \(k\) vecinos más cercanos es la existencia de un espacio de características en el cual observaciones similares se encuentran en proximidad.
A pesar de su simplicidad aparente, esta suposición plantea desafíos importantes. En primer lugar podemos plantearnos la elección adecuada del espacio de características, es decir, en la correcta selección de la métrica que definirá la similitud. No existe una fórmula mágica para determinar, a partir de un conjunto de datos dado, cuál es la métrica óptima a utilizar.
El segundo desafío se relaciona con la noción de “cercanía”. ¿Cómo definimos la cercanía entre dos observaciones? ¿Cuál es el volumen máximo del espacio dentro del cual consideramos que dos observaciones están cerca, y más allá del cual las consideramos distantes?
A pesar de estas cuestiones aparentemente simples, el método de los \(k\) vecinos más cercanos se ha demostrado efectivo en una amplia variedad de aplicaciones, tanto en clasificación como en regresión. En este apartado, exploraremos cómo funciona este algoritmo, cómo ajustar sus hiperparámetros y cómo aplicarlo de manera efectiva en situaciones del mundo real. A pesar de su simplicidad conceptual, los \(k\) vecinos más cercanos siguen siendo una herramienta valiosa en el repertorio de ML.
El algoritmo de los \(k\) vecinos más cercanos es un sencillo método de clasificación y regresión. En primer lugar, se eligen los hiperparámetros del modelo:
la métrica,
el número de vecinos, \(k\).
En los ejemplos siguientes, se podrá observar que es habitual realizar pruebas con diferentes configuraciones de hiperparámetros para identificar cuáles son los más apropiados. El principio fundamental del algoritmo implica obtener los \(k\) vecinos más cercanos para cada observación en la muestra de datos. En un problema de clasificación, el algoritmo devolverá la clase que predomina entre los \(k\) vecinos, es decir, la moda de la variable objetivo. En cambio, en un problema de regresión, el algoritmo proporcionará la media de la variable respuesta de los \(k\) vecinos más cercanos.
Para determinar el valor óptimo de \(k\) en función de los datos, ejecutamos el algoritmo \(k-NN\) múltiples veces, utilizando distintos valores de \(k\), y seleccionamos aquel valor que minimice la tasa de errores, al mismo tiempo que preserva la capacidad del algoritmo para realizar predicciones precisas en datos no vistos previamente. Es importante destacar que valores muy bajos de \(k\) pueden generar inestabilidad en el algoritmo, mientras que valores extremadamente altos de \(k\) pueden aumentar el error en las predicciones. En este proceso, se busca el equilibrio adecuado que optimice el rendimiento del algoritmo en cada situación.
NotaVentajas
Sencillez y facilidad de implementación: Es uno de los algoritmos más sencillos y fáciles de entender en ML. No requiere suposiciones complejas o entrenamientos prolongados.
Versatilidad: Se puede aplicar tanto a problemas de clasificación como de regresión.
No paramétrico: A diferencia de algunos algoritmos que asumen una distribución específica de los datos, \(k-NN\) no hace suposiciones sobre la forma de los datos, lo que lo hace adecuado para una amplia gama de situaciones.
Adaptabilidad a datos cambiantes: Puede adaptarse a cambios en los datos sin necesidad de volver a entrenar el modelo por completo, lo que lo hace útil en escenarios de datos en constante evolución. Ver Capítulo 9.
Interpretabilidad: Las predicciones de k-NN se basan en la observación de datos cercanos, lo que facilita la interpretación de las decisiones del modelo.
Robustez frente al ruido: Puede manejar datos con ruido, que no se adaptan a la distribución de la mayoría de los datos, sin un gran impacto en su rendimiento.
Facilidad para multiclases: Es aplicable a problemas de clasificación con múltiples clases sin necesidad de modificaciones significativas.
NotaDesventajas
Sensibilidad a la elección de\(k\): La elección del hiperparámetro que determina el número de vecinos puede ser crítica. Un valor demasiado pequeño puede hacer que el modelo sea sensible al ruido en los datos y cause sobreajuste, mientras que un valor demasiado grande puede hacer que el modelo sea demasiado suave y cause subajuste.
Sensibilidad a la elección de la métrica de distancia: La elección de la métrica de distancia es crucial y puede tener un impacto significativo en el rendimiento del algoritmo. No existe una métrica única que funcione para todos los problemas, y la elección de la métrica adecuada puede ser un desafío. Es muy probable que el científico de datos deba programar la métrica más adecuada para cada caso particular.
Coste computacional: En conjuntos de datos grandes, calcular las distancias entre una nueva observación y todos los puntos de datos en el conjunto de entrenamiento puede ser computacionalmente costoso y lento. Esto limita su eficiencia en aplicaciones con grandes volúmenes de datos.
Incapacidad para capturar relaciones complejas: \(k-NN\) es un algoritmo simple que no puede capturar relaciones complejas entre las características, como lo harían modelos más avanzados. No es adecuado para problemas con patrones no lineales.
Problemas en datos desbalanceados: En problemas de clasificación con clases desequilibradas, donde una clase tiene muchas más muestras que otra, \(k-NN\) tiende a sesgarse hacia la clase mayoritaria y puede no detectar bien la clase minoritaria.
Aplicamos el algoritmo a nuestro problema de los datos bancarios.
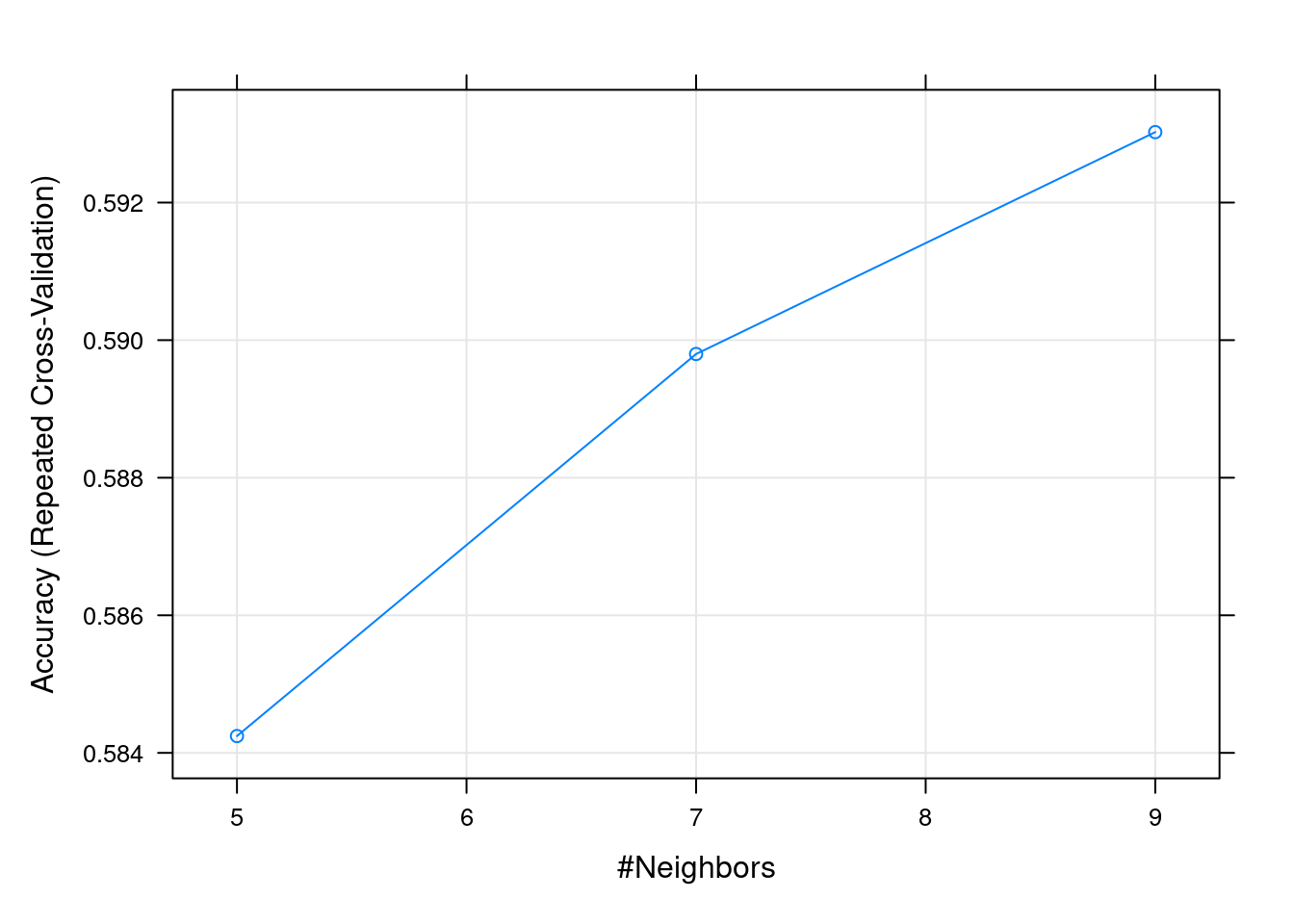
k-Nearest Neighbors
5581 samples
4 predictor
2 classes: 'no', 'yes'
No pre-processing
Resampling: Cross-Validated (10 fold, repeated 3 times)
Summary of sample sizes: 5023, 5023, 5023, 5022, 5023, 5023, ...
Resampling results across tuning parameters:
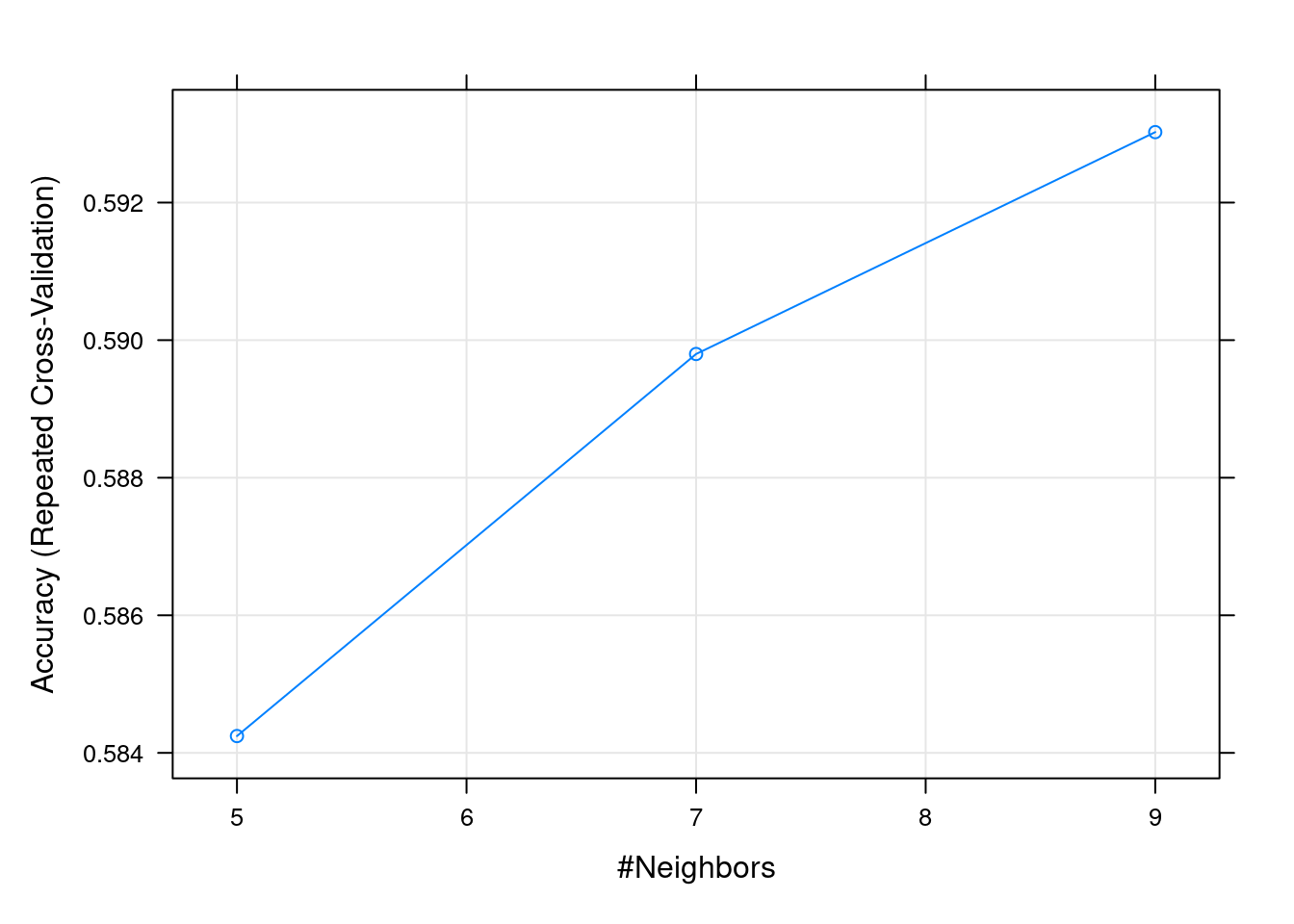
k Accuracy Kappa
5 0.5842447 0.1655072
7 0.5897985 0.1765086
9 0.5930238 0.1835312
Accuracy was used to select the optimal model using the largest value.
The final value used for the model was k = 9.
plot(fit.knn)
Según el gráfico anterior, podríamos elegir el mejor valor de \(k\), si bien puede observarse pocas diferencias entre las diferentes opciones.
A continuación, obtenemos la predicción para el conjunto de datos de prueba e imprimimos la matriz de confusión.
Confusion Matrix and Statistics
Reference
Prediction no yes
no 900 578
yes 568 744
Accuracy : 0.5892
95% CI : (0.5707, 0.6076)
No Information Rate : 0.5262
P-Value [Acc > NIR] : 1.216e-11
Kappa : 0.1759
Mcnemar's Test P-Value : 0.7903
Sensitivity : 0.5628
Specificity : 0.6131
Pos Pred Value : 0.5671
Neg Pred Value : 0.6089
Prevalence : 0.4738
Detection Rate : 0.2667
Detection Prevalence : 0.4703
Balanced Accuracy : 0.5879
'Positive' Class : yes
PrecauciónTarea
En el código anterior, ¿Porqué se calcula la media y la varianza de la variable balance?
7.3 Grid search
La “grid search” (búsqueda en cuadrícula) es una técnica comúnmente utilizada en ML para encontrar los mejores hiperparámetros para un modelo. Tal y como vimos en el Capítulo 5, los hiperparámetros son configuraciones que no se aprenden del conjunto de datos, sino que se establecen antes de entrenar el modelo. Estos hiperparámetros pueden incluir aspectos como la tasa de aprendizaje, la profundidad máxima de un árbol de decisión, el número de vecinos en el algoritmo de \(k-NN\) y otros ajustes que afectan cómo se entrena y se ajusta el modelo.
La grid search consiste en definir una “cuadrícula” de posibles valores para los hiperparámetros que se desean optimizar. Por ejemplo, si se está trabajando con un modelo de máquinas de vectores de soporte (SVM), se podría tener una cuadrícula para los hiperparámetros de la función kernel y el hiperparámetro de regularización. Luego, se entrena y evalúa el modelo utilizando todas las combinaciones posibles de valores en la cuadrícula.
El objetivo es encontrar la combinación de hiperparámetros que produce el mejor rendimiento del modelo según una métrica específica, como precisión, exactitud, \(F1\)-score, error cuadrático medio, etc.
ImportantePara recordar
La búsqueda en cuadrícula es un enfoque sistemático y automatizado que ahorra tiempo en comparación con probar manualmente diferentes combinaciones de hiperparámetros.
Si se tiene una cuadrícula de búsqueda para dos hiperparámetros, cada uno con tres valores posibles, se probarían un total de nueve combinaciones diferentes (3x3). El proceso de grid search evaluaría el rendimiento del modelo en nueve configuraciones distintas y seleccionaría aquella que obtiene los mejores resultados según la métrica definida.
Es importante mencionar exiten otras técnicas para la selección de hiperparámetros. Otros métodos, como la búsqueda aleatoria (“random search”), la optimización bayesiana y técnicas más avanzadas, también se utilizan en la selección de hiperparámetros en función de la complejidad y los recursos (computacionales, personales, temporales, etc) disponibles para la tarea.
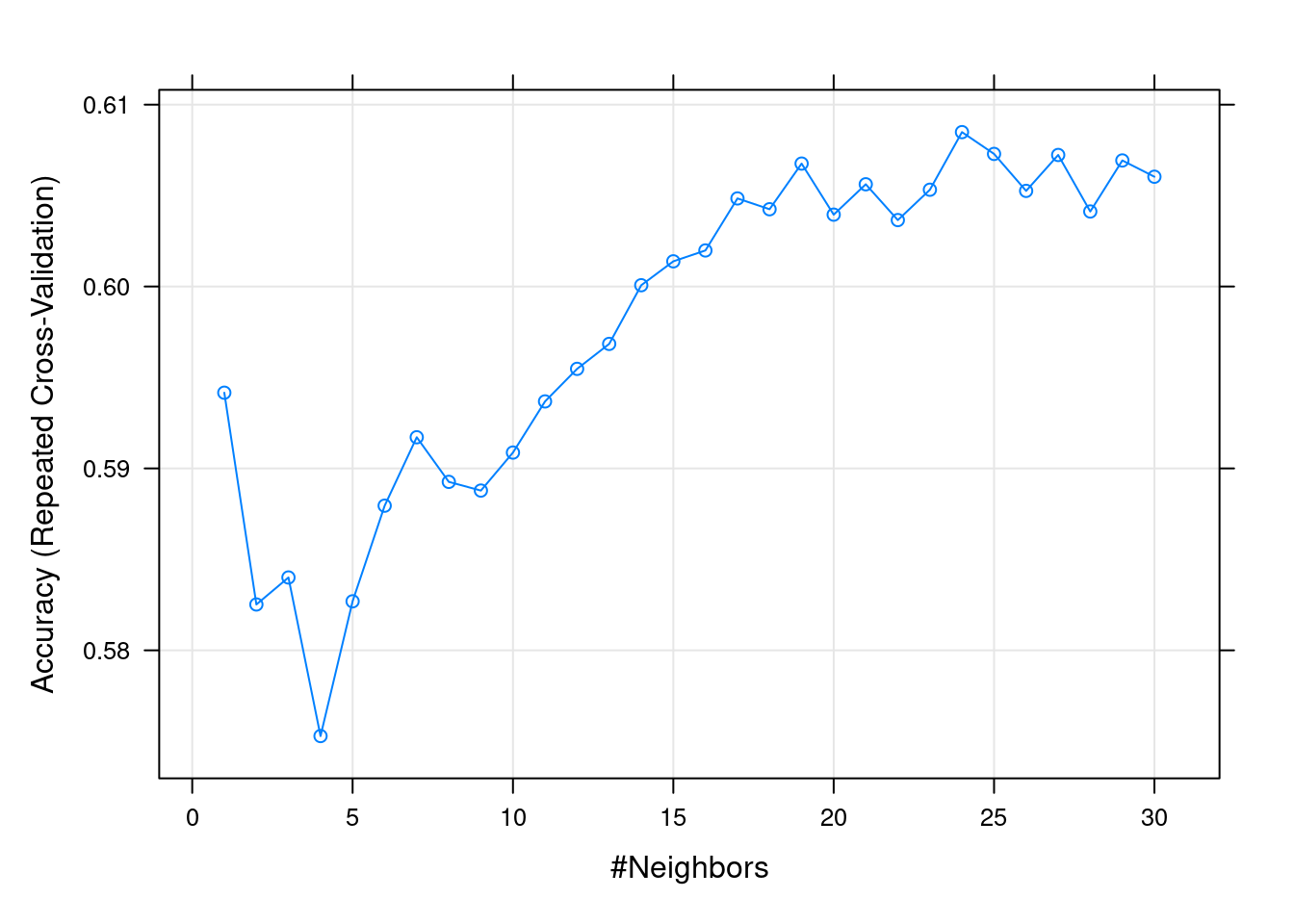
En el ejemplo que nos ocupa, buscamos el mejor valor del hiperparámetro \(k\) entre \(1\) y \(30\):
k-Nearest Neighbors
5581 samples
4 predictor
2 classes: 'no', 'yes'
No pre-processing
Resampling: Cross-Validated (10 fold, repeated 3 times)
Summary of sample sizes: 5023, 5024, 5024, 5023, 5023, 5023, ...
Resampling results across tuning parameters:
k Accuracy Kappa
1 0.5941624 0.1869556
2 0.5825199 0.1634009
3 0.5840047 0.1656685
4 0.5752866 0.1478903
5 0.5826935 0.1625400
6 0.5879512 0.1727362
7 0.5917138 0.1802904
8 0.5892608 0.1756808
9 0.5887821 0.1747647
10 0.5908722 0.1789482
11 0.5936836 0.1845274
12 0.5954742 0.1880368
13 0.5968459 0.1908545
14 0.6000722 0.1972805
15 0.6013864 0.1999850
16 0.6019835 0.2010479
17 0.6048438 0.2067664
18 0.6042492 0.2055601
19 0.6067594 0.2104826
20 0.6039522 0.2049647
21 0.6056218 0.2082912
22 0.6036533 0.2040438
23 0.6053225 0.2074437
24 0.6084891 0.2139799
25 0.6072912 0.2116389
26 0.6052587 0.2074789
27 0.6072328 0.2115650
28 0.6041256 0.2050824
29 0.6069315 0.2107200
30 0.6060367 0.2089063
Accuracy was used to select the optimal model using the largest value.
The final value used for the model was k = 24.
plot(fit.knn)
Encontramos óptimo \(k= 24\), el número de instancias más cercanas que hay que recopilar para hacer una predicción óptima. Ahora, usamos el modelo ajustado para predecir la clase para nuestro conjunto de prueba, e imprimir la matriz de confusión:
Confusion Matrix and Statistics
Reference
Prediction no yes
no 1028 603
yes 440 719
Accuracy : 0.6262
95% CI : (0.6079, 0.6442)
No Information Rate : 0.5262
P-Value [Acc > NIR] : < 2.2e-16
Kappa : 0.2457
Mcnemar's Test P-Value : 5.271e-07
Sensitivity : 0.5439
Specificity : 0.7003
Pos Pred Value : 0.6204
Neg Pred Value : 0.6303
Prevalence : 0.4738
Detection Rate : 0.2577
Detection Prevalence : 0.4154
Balanced Accuracy : 0.6221
'Positive' Class : yes
PrecauciónMétrica de disimilaridad
Hasta ahora hemos empleado la distancia Euclídea, pero ¿es lo más adecuado en este caso? Averigua cómo modificar este hiperparámetro del modelo y estudia qué consecuencias tiene.
7.4 Árboles de decisión
Los Árboles de Decisión (DT, de DecisionTree en inglés) son algoritmos de ML basados en la información. Este tipo de algoritmos determinan qué variables explicativas proporcionan la mayor parte de la información (ganancia de información) para medir la variable objetivo y hacen predicciones probando secuencialmente las características en orden a su información (Kelleher y Tierney 2018).
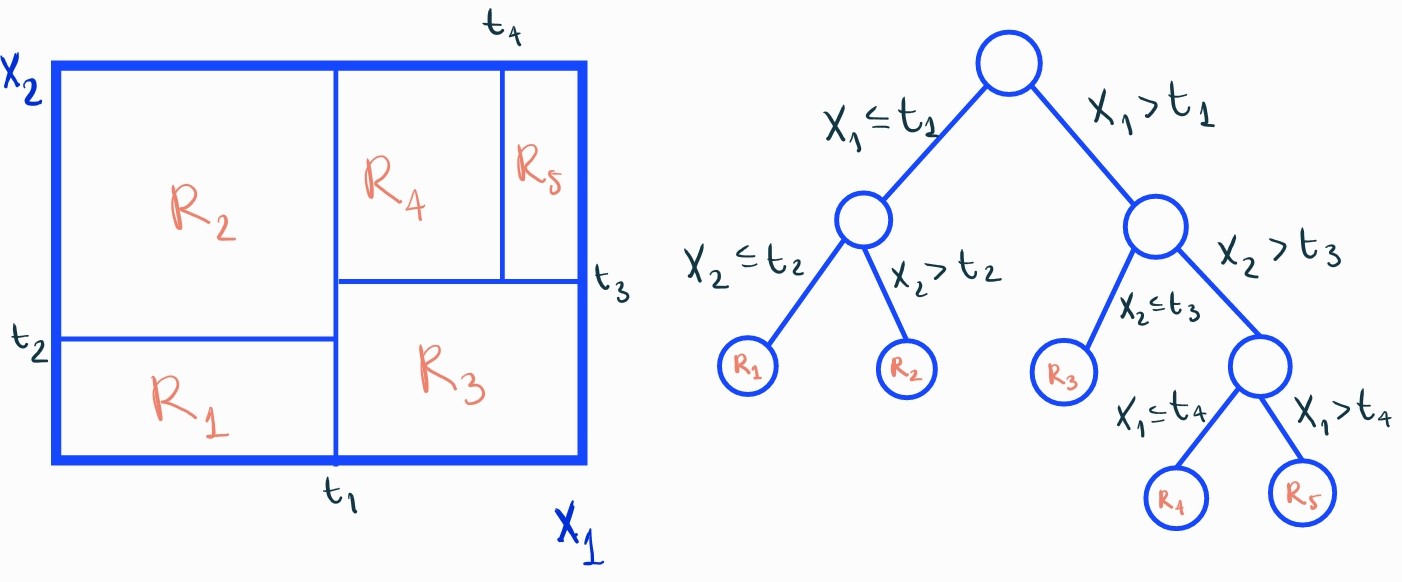
Un DT se crea mediante un proceso de particionamiento recursivo del espacio de las variables explicativas y asignando una predicción en cada una de las regiones (hojas) resultantes. Es decir, va seleccionando variables y cortes en sus valores y con ello crea una rama del árbol con dos nodos hijos. Para una mejor comprensión de la estructura de un árbol, necesitamos definir los siguientes conceptos:
Nodo raíz: es el nodo origen, inicialmente todas las observaciones forman parte de este nodo.
Nodos internos: son los nodos que se crean al definir reglas sobre una variable explicativa.
Nodos hojas: son los nodos terminales del árbol.
En la siguiente imagen se puede apreciar el funcionamiento de un árbol de decisión.
Una vez obtenidas las distintas regiones \(R_h\), el modelo sería \[f(\mathbf{x})=\sum_{h=1}^{H}g_{h}I(\mathbf{x}\in R_{h})\] siendo \(I(\mathbf{x}\in R_{h})\) la función indicatriz que vale \(1\) si la observación \(\mathbf{x}\) pertenece a la región \(R_{h}\) y \(g_{h}\) la predicción (el valor de la variable respuesta) que se le asignará a dicha observación. En un problema de regresión \(g_{h}\) será la media de la variable respuesta \(y\) en dicha región. En un problema de clasificación, será la proporción de las clases.
Nótese que, habitualmente, favorecemos árboles que empleen el menor número de preguntas posible, lo que significa que buscamos árboles poco profundos. La construcción de árboles con poca profundidad implica que las características más informativas, es decir, aquellas que mejor distinguen entre observaciones con diferentes valores en la variable objetivo, deben ubicarse en la parte superior del árbol. Para llevar a cabo esta tarea, es fundamental contar con una métrica formal que evalúe cuán efectivamente una variable discrimina entre los niveles de la variable objetivo.
TipEntropía
La entropía es una medida teórica de la “incertidumbre” contenida en un conjunto de datos, debido a la presencia de más de una posibilidad de clasificación.
La entropía es una medida de la impureza (heterogeneidad), de los datos. Desde un punto de vista formal, la entropía de un conjunto de \(N\) valores distintos que son igualmente probables es el menor número de preguntas de tipo sí/no necesarias para determinar un valor desconocido extraído de las \(N\) posibilidades:
\[\text{Entropía}=-\sum_{i=1}^N p_i log_2p_i\]
Podemos ver que la entropía, por definición, toma valores mayores o iguales a cero. Su valor máximo se alcanza cuando las observaciones se distribuyen equitativamente entre las \(N\) posibles clases. La entropía toma su valor mínimo si, y sólo si, todas las observaciones están en la misma clase. Supongamos dos clases, esto es \(N=2\). En ese caso, consideramos la probabilidad de clase \(1\) y la probabilidad de clase \(2\) (\(p_1\) y \(p_2\) respectivamente). Si todas las observaciones están en la misma clase (por ejemplo, la clase \(1\)), entonces: \(p_1=1\), \(p_2=0\). Y así:
El algoritmo de DT basado en ganancia de la información sería como sigue:
Encontrar la mejor variable y su mejor corte. Para ello, dado el conjunto de datos \(\mathcal{D}=\{(\mathbf{x}_i,y_i)\}_{i=1}^{n}\) se evalúan las posibles divisiones basadas en las variables \(\mathbf{x}_j\) y se selecciona aquella que maximiza la reducción de impureza en los nodos. Esto es:
Para cada variable \(\mathbf{x}_j\) en el dataset, considerar el conjunto de puntos de corte \(S_j\) (la media entre valores consecutivos para datos continuos)
Para cada punto de corte \(s \in S_j\), calcular la impureza resultante tras la división:
Dividir el dataset en dos subconjuntos (nodos hoja): \[\mathcal{D}_L= \{ (\mathbf{x}_i,y_i) \in \mathcal{D} | x_{ij} < s \}\]\[\mathcal{D}_R= \{ (\mathbf{x}_i,y_i) \in \mathcal{D} | x_{ij} \geq s \}\]
Calcular la impureza de cada subconjunto con el criterio seleccionado (por ejemplo, entropía) Calcular la reducción de impureza: \[\Delta I = I(\mathcal{D})-\left( \frac{|\mathcal{D}_L|}{|\mathcal{D}|}I(\mathcal{D}_L) + \frac{|\mathcal{D}_R|}{|\mathcal{D}|}I(\mathcal{D}_R)\right)\] donde \(I(\mathcal{D})\) es la impureza antes de dividir (nodo padre) e \(I(\mathcal{D}_L)\), \(I(\mathcal{D}_R)\) son las impurezas después de dividir (nodos hijo).
Seleccionar la mejor variable y punto de corte \((\mathbf{x}_{j}^{*},s^{*})\). La combinación que maximiza \(\Delta I\) se elige como la mejor división en ese nodo.
Repetir recursivamente en cada nodo hijo hasta alcanzar un criterio de parada
Es decir, la idea es emplear los valores de las características medidas sobre las observaciones para dividir el conjunto inicial de datos en subconjuntos más pequeños repetidamente, hasta que la entropía de cada uno de los subconjuntos sea cero (o pequeña, menor que un umbral). Para cada partición, la entropía media de los subconjuntos resultantes debería ser menor que la del conjunto anterior.
En términos generales, la ganancia de información se define como la diferencia entre la entropía anterior y la nueva entropía. Por lo tanto, para cada nodo en el árbol, se elige para la división aquella variable que maximiza la ganancia de información.
NotaÍndice de Gini
Existen otras métricas posibles para medir la calidad de la división. Por ejemplo, el índice de Gini:
\[Gini=\sum_{i=1}^N p_i (1-p_i)\] siendo \(p_i\) la proporción de ejemplos de la clase \(i\). También sirve para cuantificar lo “mezcladas” que están las clases dentro de un nodo. Cuanto menor sea el índice de Gini, mejor es la pureza del nodo.
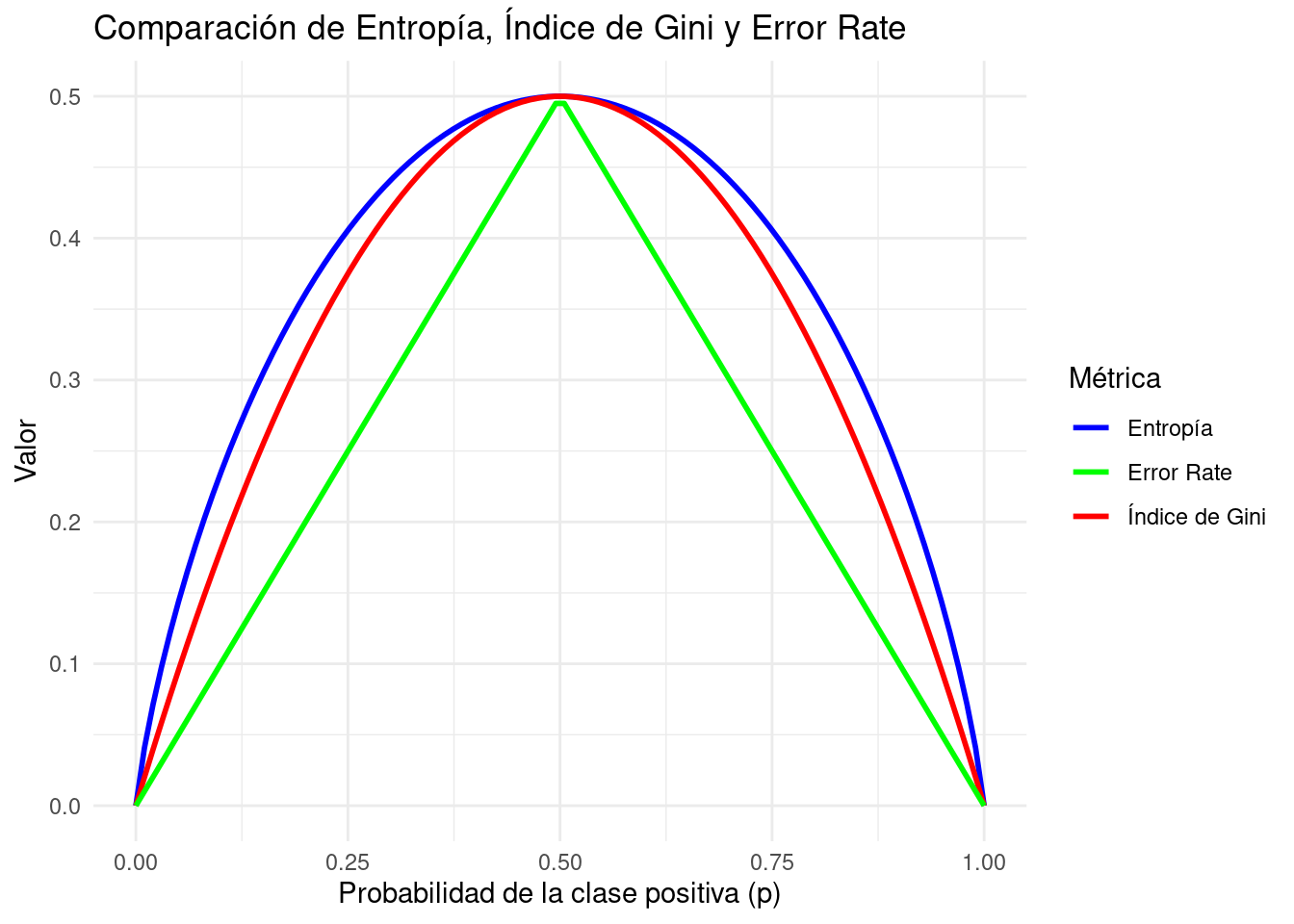
En el siguiente gráfico se muestra el comportamiento de la entropía, del índice de Gini y del ratio de error (nótese que la entropía ha sido dividida entre 2 para facilitar la visualización).
library(ggplot2)# Secuencia de probabilidadesp <-seq(0, 1, length.out =100)# Cálculo de las medidasentropy <--p *log2(p) - (1- p) *log2(1- p)entropy[is.na(entropy)] <-0# log(0) da NaNgini <-2* p * (1- p)error_rate <-pmin(p, 1- p)df <-data.frame(p, Entropy = entropy, Gini = gini, ErrorRate = error_rate)# Gáficoggplot(df, aes(x = p)) +geom_line(aes(y = Entropy/2, color ="Entropía"), linewidth =1) +geom_line(aes(y = Gini, color ="Índice de Gini"), linewidth =1) +geom_line(aes(y = ErrorRate, color ="Error Rate"), linewidth =1) +labs(title ="Comparación de Entropía, Índice de Gini y Error Rate",x ="Probabilidad de la clase positiva (p)",y ="Valor",color ="Métrica") +theme_minimal() +scale_color_manual(values =c("Entropía"="blue", "Índice de Gini"="red", "Error Rate"="green"))
PrecauciónTarea
¿Por qué crees que se utiliza la entropía o el índice de Gini y no se usa el ratio de error como medida para la construcción del árbol?
En el caso de árboles de regresión, donde la variable objetivo es cuantitativa, se define la reducción de la varianza como la disminución total de la varianza de la variable objetivo como resultado de la división del conjunto total en dos subconjuntos.
Existen varios criterios para detener la ejecución de este algoritmo de división. En un caso extremo podemos no detenerlo hasta que el error de clasificación sea \(0\). En dicho caso posiblemente cometamos errores de sobreajuste. Es decir, seleccionar divisiones sobre las variables hasta que todas las observaciones de la muestra de entrenamiento están perfectamente clasificadas funcionará bien, pero sólo sobre las observaciones de entrenamiento. Cuando ese mismo criterio se aplique a nuevas observaciones el resultado será, probablemente, mucho peor. En ocasiones se detiene el proceso de particionado cuando la profundidad del árbol supera un umbral (esto es, cuando se han empleado un número determinado de preguntas sobre las variables explicativas), o bien cuando el número de observaciones en el nodo a dividir es menor que un valor prefijado. También se puede evaluar si la reducción del criterio escogido (índice de Gini o entropía) es demasiado pequeña.
Poda del árbol de decisión
Los árboles de decisión tienden a sobreajustarse a los datos de entrenamiento, por lo que se aplica una técnica de poda para mejorar la generalización. Existen dos enfoques principales:
Poda pre-pruning (previa a la construcción completa del árbol): Se establece un criterio de parada como la profundidad máxima, el número mínimo de muestras en un nodo o un umbral mínimo de ganancia de información para realizar una división.
Poda post-pruning (posterior a la construcción del árbol completo): Se construye el árbol hasta su máxima profundidad y luego se eliminan nodos internos si su eliminación mejora la métrica de validación (por ejemplo, validación cruzada). Una técnica común es la poda por reducción de error, donde se evalúa si eliminar un nodo interno mejora el rendimiento del modelo. Esta poda se puede formalizar mediante la minimización de una función de coste regularizada: \[C(T)=C_t(T) + \alpha |T|\] donde \(T\subset T_{full}\) es cualquier árbol que se pueda lograr podando el árbol completo \(T_{full}\), \(|T|\) es el número de nodos terminales en \(T\), \(C_t(T)\) es el error en el entrenamiento del subárbol \(T\) y \(\alpha\) es un hiperparámetro que controla la penalización por la complejidad del modelo. Este hiperpárametro \(\alpha\) rige el equilibrio entre el tamaño del árbol y su ajuste a los datos. Valores grandes, dan como resultados árboles pequeños y, por otro lado, valores pequeños de \(\alpha\) dan lugar a árboles de mayor tamaño. Con \(\alpha=0\), \(T=T_{full}\).
ImportantePara recordar
La salida de un DT es un conjunto de reglas que se puede representar como un árbol donde cada nodo representa una decisión binaria.
NotaVentajas
7.5 Ejemplo: Construcción de un árbol de decisión usando entropía de Shannon
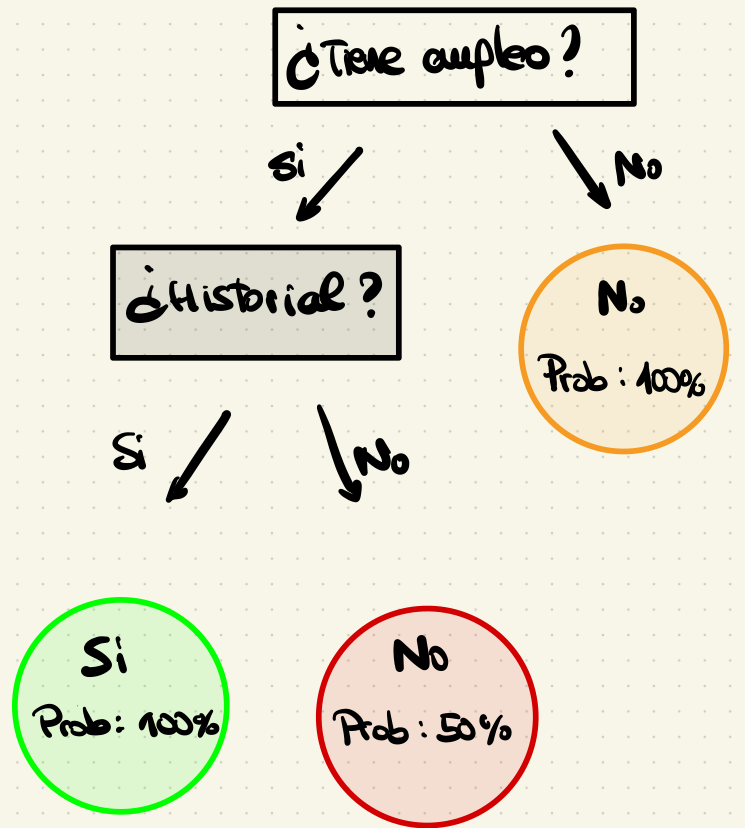
Supongamos que trabajamos en un banco y queremos predecir si un cliente pagará o no un préstamo. Para ello, tenemos un conjunto de datos con dos características binarias:
Tiene empleo (Sí/No)
Historial crediticio positivo (Sí/No)
Variable objetivo Paga préstamo (Sí/No)
Datos de clientes y su historial de préstamos
ID
Tiene empleo
Historial crediticio
Paga préstamo
1
Sí
Sí
Sí
2
Sí
No
Sí
3
No
Sí
No
4
No
No
No
5
Sí
Sí
Sí
6
No
Sí
No
7
Sí
No
No
8
No
No
No
Queremos construir un árbol de decisión utilizando la entropía de Shannon para seleccionar la mejor variable en cada división.
Dado que hay ganancia de información, hacemos la división.
El árbol resultante es:
Así, tenemos 3 nodos terminales. Dos de ellos son puros y otro está en la situación de máxima incertidumbre. No obstante, no disponemos de más variables para seguir construyendo el árbol.
A continuación aplicamos el método de DT al conjunto de datos bank.
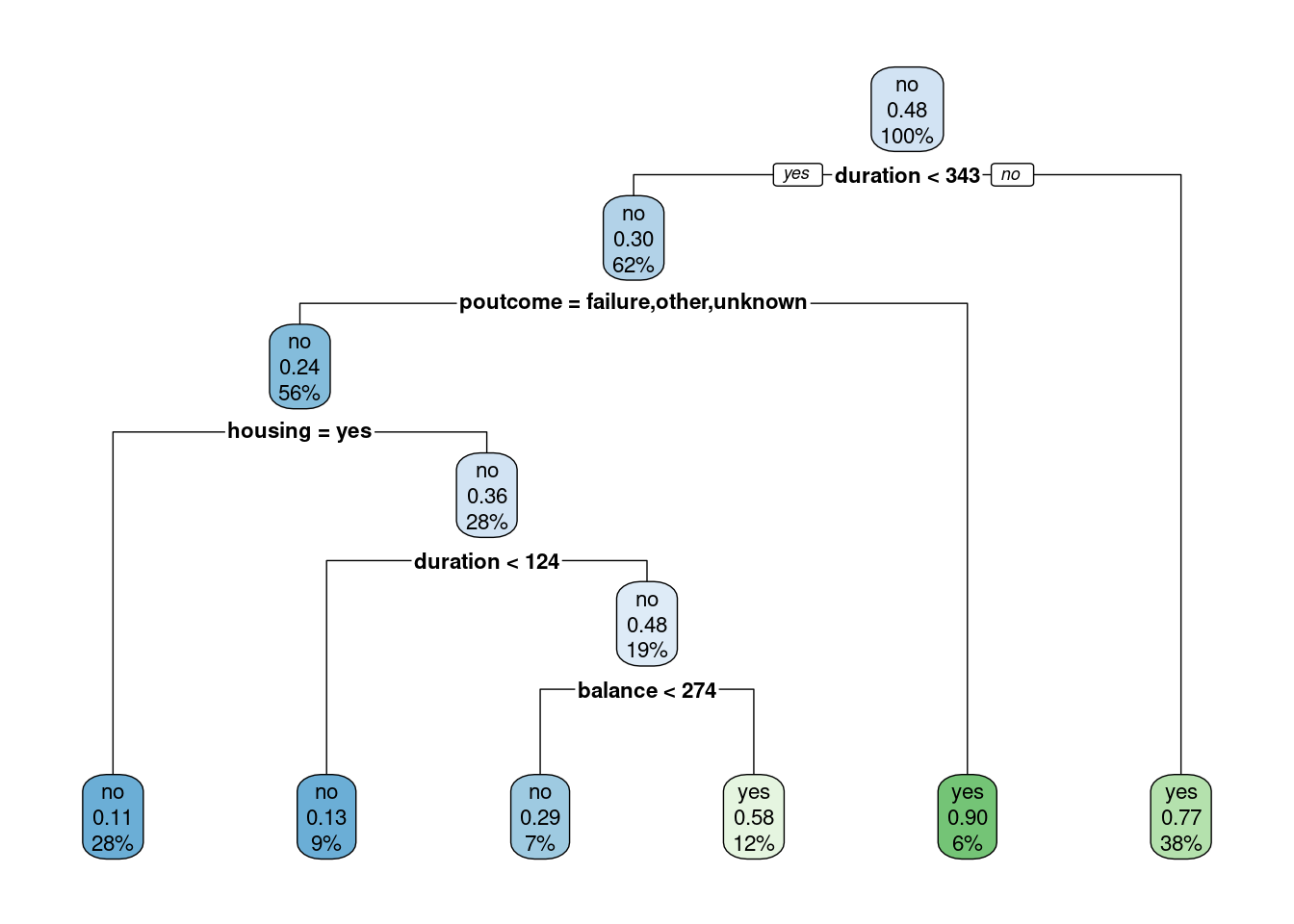
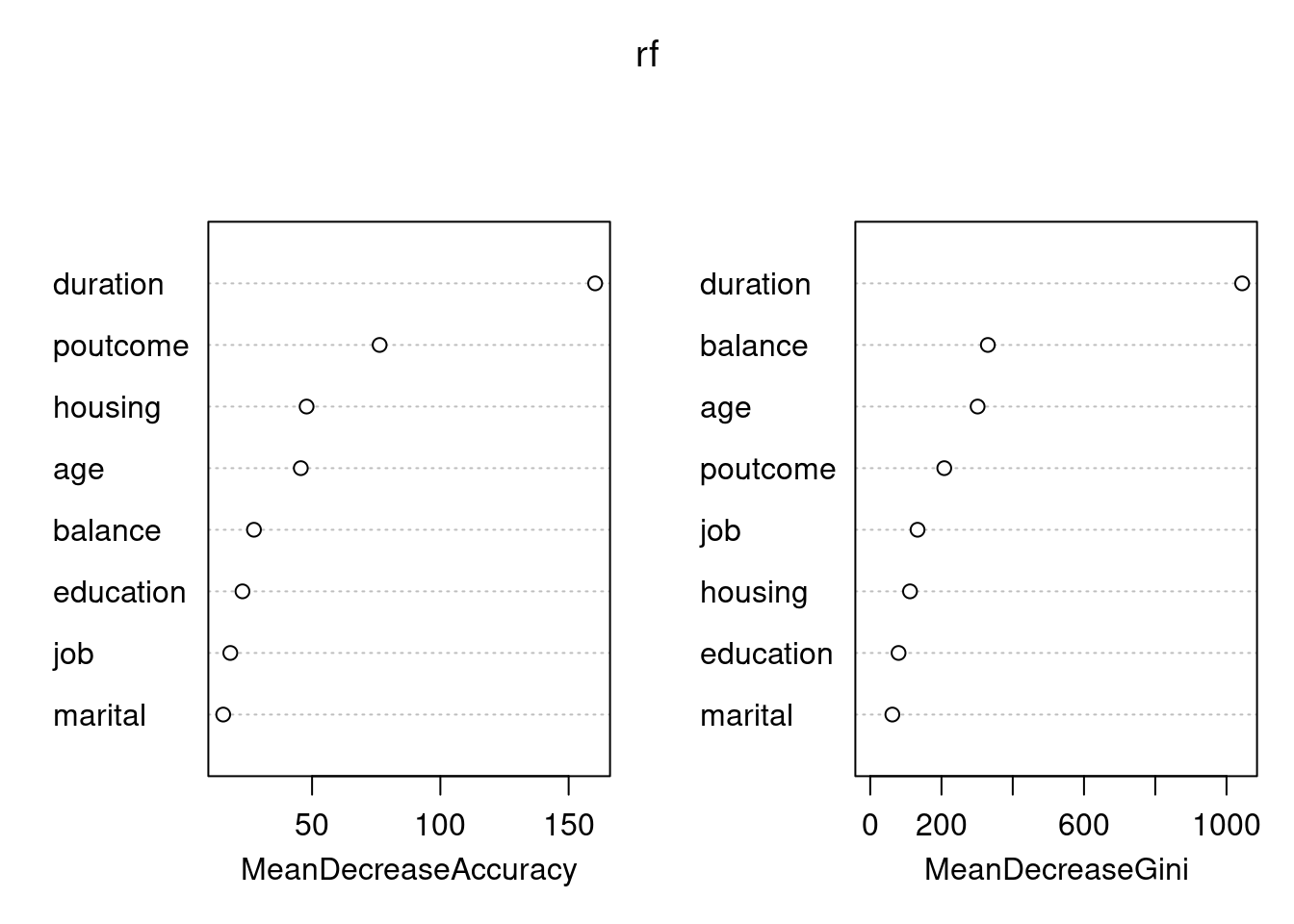
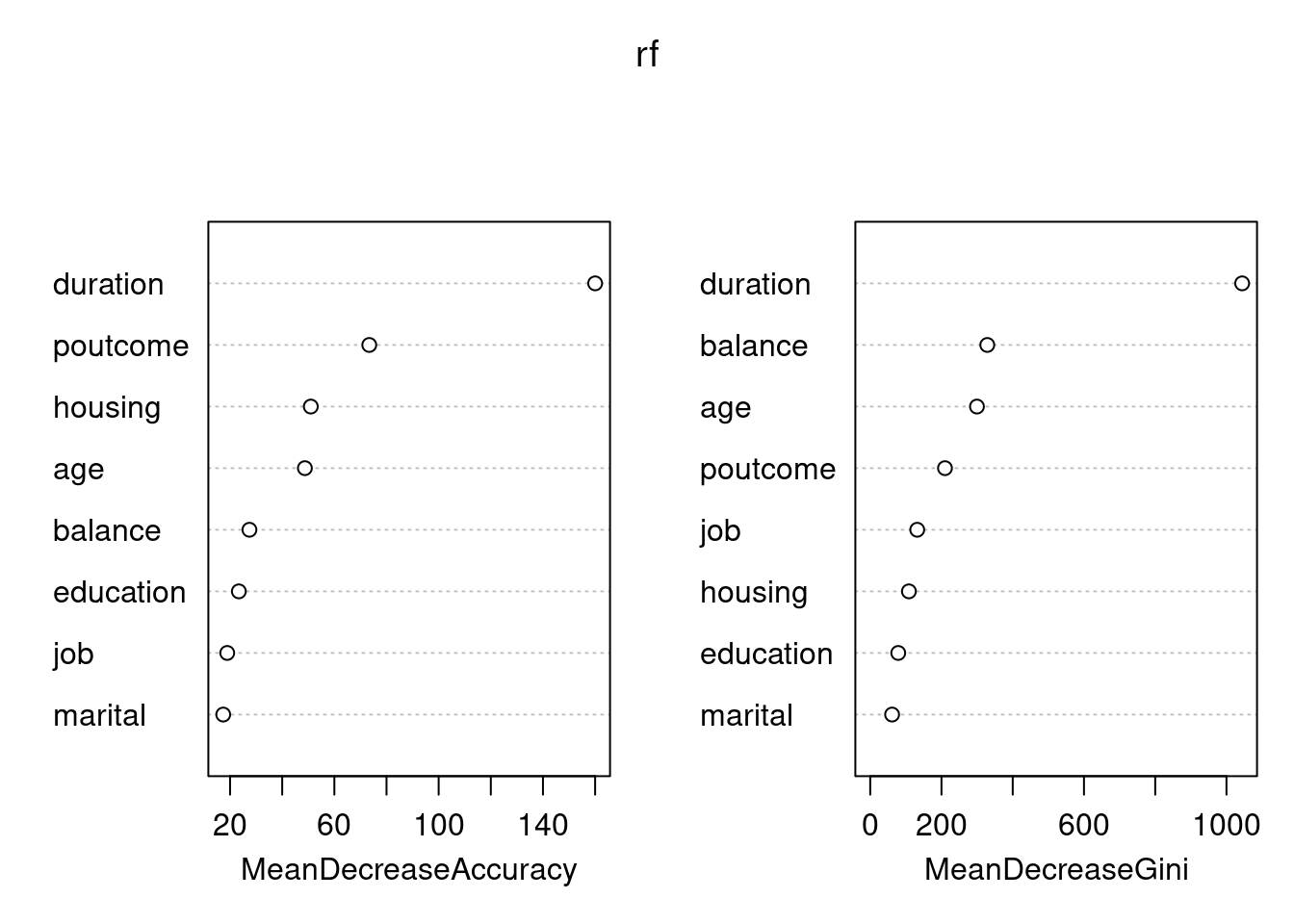
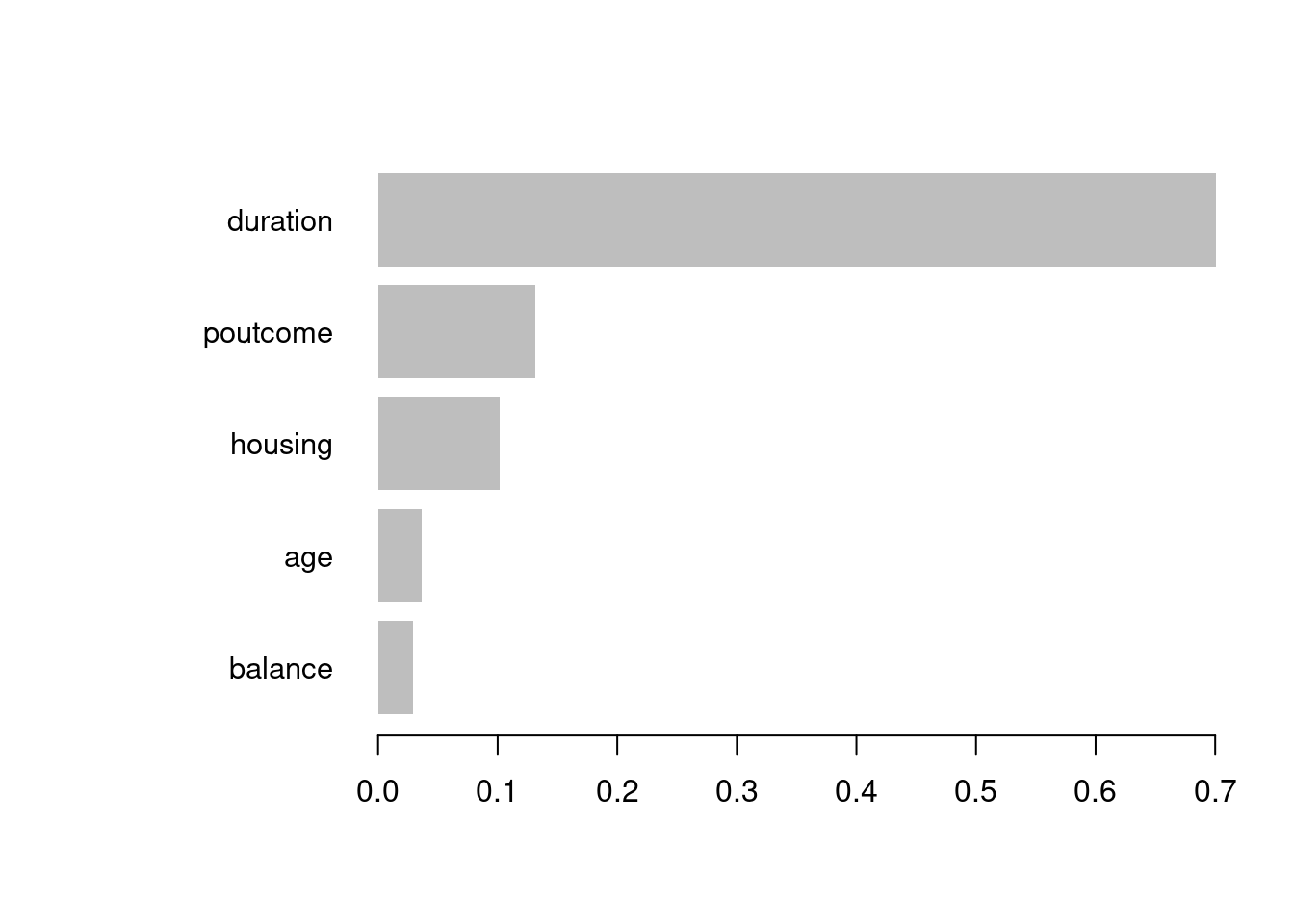
library(rpart)library(rpart.plot)set.seed(128)df <- bank.train %>% dplyr::select(age,job,housing,marital,education,duration,poutcome,balance,deposit)fit.dt <-rpart(deposit~., data = df, method ='class')rpart.plot(fit.dt, extra =106)
Como podemos observar, la interpretación del modelo de DT es muy sencilla. Todas las observaciones están en el primer nodo (el nodo raíz). Sin realizar “preguntas” sobre las variables explicativas, el nodo es de clase “no” con una probabilidad de “yes” del \(0.48\).
La primera variable de interés es la duration:
Si el valor es menor que \(343\) (este valor es aprendido por el algoritmo y puede ser difícil de interpretar) entonces la observación va hacia la rama izquierda, cayendo en un nodo dónde la probabilidad de “yes” ha disminuido a \(0.30\). En ese nodo aparecen el \(62\%\) de todas las observaciones del conjunto de entrenamiento.
Por contra, si el valor de durationes mayor que \(343\) entonces la observación va hacia la rama derecha, cayendo en un nodo dónde la probabilidad de “yes” ha aumentado a \(0.77\). En ese nodo aparecen el \(38\%\) de todas las observaciones del conjunto de entrenamiento. Además, fíjate que se trata de un nodo terminal.
PrecauciónTarea
No se están teniendo en cuenta todas las variables disponibles en el conjunto de datos. El objetivo de la mayoría de los ejercicios de estos apuntes es mostrar a los alumnos cómo funcionan los métodos de ML. Si tu objetivo es obtener la más alta precisión, o el menor error, entonces te animamos a que intentes construir mejores modelos con, probablemente, más y mejores variables.
Podemos interpretar el resto de los nodos terminales del ejemplo como sigue:
Nodo
Descripción
Tamaño relativo (%)
Prob(“yes”)
1
duration < 343, poutcome=failure,other or unkonwn, housing=yes
26.0
0.11
2
duration < 124, poutcome=failure,other or unkonwn, housing=yes
9.0
0.13
3
124<=duration < 343, poutcome=failure,other or unkonwn, housing=yes, balance<274
7.0
0.29
4
124<=duration < 343, poutcome=failure,other or unkonwn, housing=yes, balance>=274
12.0
0.58
5
duration < 343, poutcome=success
6.0
0.90
6
duration>343
38.0
0.77
Usamos el modelo de DT para hacer predicciones:
# sobre la partición de entrenamientoprediction <-predict(fit.dt, df, type ='class')cf <-confusionMatrix(prediction, as.factor(df$deposit),positive="yes")print(cf)
Confusion Matrix and Statistics
Reference
Prediction no yes
no 2090 349
yes 818 2324
Accuracy : 0.7909
95% CI : (0.78, 0.8015)
No Information Rate : 0.5211
P-Value [Acc > NIR] : < 2.2e-16
Kappa : 0.584
Mcnemar's Test P-Value : < 2.2e-16
Sensitivity : 0.8694
Specificity : 0.7187
Pos Pred Value : 0.7397
Neg Pred Value : 0.8569
Prevalence : 0.4789
Detection Rate : 0.4164
Detection Prevalence : 0.5630
Balanced Accuracy : 0.7941
'Positive' Class : yes
# sobre la partición de pruebaprediction.dt <-predict(fit.dt, df.test, type ='prob')[,2]clase.pred=ifelse(prediction.dt>0.5,"yes","no")cf <-confusionMatrix(as.factor(clase.pred), as.factor(df.test$deposit),positive="yes")print(cf)
Confusion Matrix and Statistics
Reference
Prediction no yes
no 1178 381
yes 290 941
Accuracy : 0.7595
95% CI : (0.7432, 0.7753)
No Information Rate : 0.5262
P-Value [Acc > NIR] : < 2.2e-16
Kappa : 0.516
Mcnemar's Test P-Value : 0.000512
Sensitivity : 0.7118
Specificity : 0.8025
Pos Pred Value : 0.7644
Neg Pred Value : 0.7556
Prevalence : 0.4738
Detection Rate : 0.3373
Detection Prevalence : 0.4412
Balanced Accuracy : 0.7571
'Positive' Class : yes
Al igual que en otros métodos de ML, podemos ajustar los hiperparámetros del modelo. En este caso ajustamos la máxima profundidad del árbol, el mínimo número de observaciones en un nodo para poder ser particionado en dos y el mínimo número de observaciones que un nodo terminal debe tener.
control <-rpart.control(minsplit =4,minbucket =round(5/3),maxdepth =3,cp =0)tune.fit <-rpart(deposit~., data = df, method ='class', control = control)rpart.plot(tune.fit, extra =106)
El nuevo modelo presenta un rendimiento ligeramente menor que el original pero respondiendo a condiciones que antes no imponíamos.
# sobre la partición de pruebaprediction <-predict(tune.fit, df.test, type ='class')cf <-confusionMatrix(prediction, as.factor(df.test$deposit),positive="yes")print(cf)
Confusion Matrix and Statistics
Reference
Prediction no yes
no 1186 393
yes 282 929
Accuracy : 0.7581
95% CI : (0.7417, 0.7739)
No Information Rate : 0.5262
P-Value [Acc > NIR] : < 2.2e-16
Kappa : 0.5128
Mcnemar's Test P-Value : 2.297e-05
Sensitivity : 0.7027
Specificity : 0.8079
Pos Pred Value : 0.7671
Neg Pred Value : 0.7511
Prevalence : 0.4738
Detection Rate : 0.3330
Detection Prevalence : 0.4341
Balanced Accuracy : 0.7553
'Positive' Class : yes
NotaVentajas
Explicabilidad: Los DT son altamente interpretables, lo que significa que sus resultados se pueden entender y comunicar de manera sencilla (ver Capítulo 9). Esto los hace ideales para tomar decisiones basadas en modelos en entornos donde se necesita entender el razonamiento detrás de las predicciones.
Compatibilidad con datos mixtos: Pueden manejar tanto variables explicativas cuantitativas como cualitativas (categóricas). Esta versatilidad les permite trabajar con una amplia variedad de tipos de datos.
Clasificación multiclase: Los DT se pueden extender de manera natural para la clasificación multiclase, lo que significa que pueden asignar observaciones a más de dos categorías.
Escalado de variables no requerido: A diferencia de algunos otros algoritmos, los DT no requieren que las variables se escalen o estandaricen antes de su uso. Esto facilita el proceso y ahorra tiempo en la preparación de datos.
Manejo de datos faltantes: Los árboles pueden manejar datos faltantes sin necesidad de preprocesamiento adicional. Tratan los valores faltantes como una categoría adicional y no descartan observaciones con datos faltantes.
Captura de no linealidades: Los DT son capaces de captar relaciones no lineales entre variables, lo que es valioso cuando las relaciones entre las características y la variable objetivo son complejas y no se ajustan bien a modelos lineales.
Robustez a valores atípicos: Los árboles son menos sensibles a valores atípicos en los datos en comparación con algunos otros algoritmos de aprendizaje automático. Los valores atípicos no tienen un impacto tan drástico en la construcción de árboles como en modelos lineales, por ejemplo.
Eficiencia: Son relativamente eficientes en términos computacionales, especialmente para conjuntos de datos de tamaño moderado. La construcción de árboles puede realizarse de manera rápida.
NotaDesventajas
Menos precisión en modelos simples: Los DT a menudo son menos precisos que métodos más avanzados, como los bosques aleatorios o las redes neuronales, especialmente cuando se trata de problemas complejos. Su simplicidad a veces limita su capacidad para modelar relaciones sofisticadas en los datos.
Sensibilidad a cambios en los datos: Pequeños cambios en los datos de entrenamiento pueden resultar en DT significativamente diferentes. Esto significa que los modelos basados en árboles son menos estables y robustos en comparación con algunos otros algoritmos. Un cambio en una observación o característica puede dar como resultado un árbol completamente diferente, lo que dificulta la confianza en las predicciones.
Propensión al sobreajuste: Los DT tienden a sobreajustarse a los datos de entrenamiento cuando se construyen demasiado profundos. Esto significa que el modelo se adapta demasiado a los datos de entrenamiento y puede no generalizar bien a nuevos datos. La elección de la profundidad óptima del árbol es crítica para evitar el sobreajuste.
Dificultad para modelar relaciones lineales: A pesar de su capacidad para capturar relaciones no lineales, los DT no son ideales para modelar relaciones lineales en los datos. En tales casos, otros modelos, como la regresión lineal, pueden ser más apropiados.
Limitación en la predicción de valores continuos: Los DT son adecuados para la clasificación, pero no son la mejor opción para la regresión o la predicción de valores continuos. Pueden proporcionar predicciones discretas, lo que puede ser una limitación en aplicaciones donde se requieren estimaciones precisas de valores numéricos.
Sensibilidad a valores atípicos: Aunque son menos sensibles a los valores atípicos que algunos otros algoritmos, los DT todavía pueden verse afectados por valores extremos en los datos, lo que puede influir en la construcción del árbol y, por lo tanto, en las predicciones.
No son óptimos para datos de alta dimensión: Para conjuntos de datos de alta dimensión, la construcción de árboles puede volverse compleja y computacionalmente costosa. En tales casos, es posible que otros algoritmos, como las máquinas de vectores soporte (SVM) o el aprendizaje profundo, sean más apropiados.
Difícil interpretación en árboles profundos: Si los árboles se construyen muy profundos, pueden volverse difíciles de interpretar y visualizar, lo que reduce su utilidad en aplicaciones que requieren explicaciones claras de las decisiones del modelo.
Si bien los DTs son una herramienta valiosa, es importante considerar estas limitaciones al seleccionar la técnica de ML adecuada para un problema específico. Las desventajas mencionadas anteriormente son algunas de las razones por las que se han desarrollado métodos más avanzados, basados en ensamblado como los Bosques Aleatorios, para abordar algunas de estas limitaciones.
7.6 Métodos de ensamblado
Los métodos de ensamblado son una potente estrategia en el campo del ML que se basa en la idea de que la unión de múltiples modelos puede mejorar significativamente el rendimiento de predicción en comparación con un solo modelo.
NotaLa unión hace la fuerza
En lugar de depender de un solo algoritmo para tomar decisiones, los métodos de ensamblado combinan las predicciones de varios modelos para obtener resultados más precisos y robustos.
Esta técnica se asemeja a la sabiduría colectiva: al reunir a un grupo diverso de personas, cada una con su conjunto único de conocimientos y perspectivas, se puede tomar una decisión más sólida y precisa. Del mismo modo, los métodos de ensamblado combinan múltiples modelos, cada uno de los cuales puede sobresalir en diferentes aspectos del problema, para lograr una predicción más precisa y confiable.
Los métodos de ensamblado se han convertido en un componente esencial en la caja de herramientas de los científicos de datos y profesionales del ML. Estas técnicas utilizan una variedad de algoritmos base, como DT, regresión logística, máquinas de vectores soporte y más, y los combinan de manera inteligente para mejorar la capacidad de generalización del modelo. En este enfoque, se pueden distinguir dos categorías principales de métodos de ensamblado:
el ensamblado de bagging
el ensamblado de boosting
7.6.1 Bagging
El ensamblado de Bagging, (de “Bootstrap Aggregating” en inglés), es una técnica de ML diseñada para mejorar la precisión y la estabilidad de los modelos predictivos. Se basa en la idea de construir múltiples modelos similares y combinar sus predicciones para obtener un resultado final más robusto y generalizable.
Ya vimos que uno de los problemas de los árboles de decisión era que son clasificadores con alta varianza. La técnica bagging nació con el objetivo de reducir dicha varianza dado que una manera de reducir la varianza de un estimador es hacer la media de varios estimadores. Se entrenan \(M\) árboles distintos \(f_m(\mathbf{x}), m=1,\dots, M\) en diferentes subconjuntos del conjunto de datos (elegidos aleatoriamente con remplazamiento) y se calcula el ensamblado total
Damos, a continuación, una explicación detallada de cómo funciona el ensamblado de Bagging:
Bootstrap: El proceso comienza dividiendo el conjunto de datos original en múltiples subconjuntos llamados conjuntos de entrenamiento, utilizando un método llamado muestreo con reemplazo. Esto significa que, en cada conjunto de entrenamiento, algunas muestras se seleccionan más de una vez, mientras que otras pueden quedar fuera. Este proceso genera múltiples conjuntos de entrenamiento, cada uno ligeramente diferente del original.
Modelo base: Luego, se entrena un modelo base, como un DT, regresión logística o cualquier otro algoritmo, en cada uno de estos conjuntos de entrenamiento. Cada modelo base se entrena en datos diferentes debido al muestreo con reemplazo, lo que da como resultado una serie de modelos que pueden variar en pequeñas diferencias.
Predicciones: Una vez que se han entrenado todos los modelos base, se utilizan para hacer predicciones individuales sobre un conjunto de datos de prueba.
Combinación: Finalmente, las predicciones de todos los modelos base se combinan para obtener una predicción agregada. En problemas de clasificación, esto suele implicar votación, donde se elige la clase con más votos, y en problemas de regresión, se calcula un promedio de las predicciones.
NotaVentajas
Reducción de la varianza: Al crear múltiples conjuntos de entrenamiento y modelos base, el ensamblado de Bagging reduce la varianza de las predicciones, lo que hace que el modelo sea más robusto y menos propenso al sobreajuste.
Mayor precisión: La combinación de múltiples modelos base suele resultar en una precisión general superior en comparación con un solo modelo.
Estabilidad: Al construir modelos ligeramente diferentes a partir de diferentes subconjuntos de datos, el ensamblado de Bagging aumenta la estabilidad y la resistencia del modelo ante datos ruidosos o atípicos.
Mayor generalización: Debido a su capacidad para reducir el sobreajuste, el ensamblado de Bagging es efectivo para problemas de alta dimensionalidad y datos con ruido.
NotaDesventajas
Mayor complejidad computacional: Debido a la necesidad de entrenar múltiples modelos base, el Bagging puede requerir más recursos computacionales y tiempo de entrenamiento en comparación con un solo modelo. Esto es especialmente relevante cuando se trabaja con conjuntos de datos grandes o algoritmos de modelo base complejos.
Menos interpretabilidad: La combinación de múltiples modelos base dificulta la interpretación del modelo global. A diferencia de modelos individuales, como DT simples, los modelos Bagging no proporcionan una descripción sencilla de cómo se llega a una decisión o predicción.
No garantiza la mejora: Aunque el Bagging suele mejorar la precisión y reducir la varianza en comparación con un solo modelo base, no garantiza un mejor rendimiento en todos los casos. En algunos conjuntos de datos o problemas, el Bagging puede no proporcionar mejoras significativas y, en raras ocasiones, podría incluso empeorar el rendimiento.
Menos efectivo con modelos base inestables: Si se utilizan modelos base que son inherentemente inestables o propensos al sobreajuste, el Bagging puede no ser tan efectivo en mejorar su rendimiento. En tales casos, otros métodos de ensamblado, como Boosting, podrían ser más apropiados.
Limitaciones en problemas de desequilibrio de clases: En problemas de clasificación con desequilibrio de clases, el Bagging puede no abordar adecuadamente el desafío de predecir clases minoritarias. En tales situaciones, se requieren técnicas específicas, como el ajuste de pesos de clase, para abordar el desequilibrio.
El algoritmo más conocido que utiliza Bagging es el “Random Forest”, que combina múltiples DTs para lograr un modelo predictivo altamente preciso.
7.6.1.1 Random Forest
Un Random Forest o Bosque Aleatorio es una potente técnica de ensamblado que utiliza un conjunto de DTs para mejorar la precisión de las predicciones. En lugar de confiar en un solo árbol, se construye un bosque compuesto por numerosos árboles individuales. La singularidad de un Bosque Aleatorio radica en cómo se crean y combinan estos árboles.
Cada árbol dentro del Bosque Aleatorio no se crea a partir de todo el conjunto de datos, sino que se entrena con un subconjunto aleatorio de variables y un conjunto de observaciones seleccionadas al azar. Este proceso de muestreo aleatorio introduce diversidad en la construcción de cada árbol, lo que ayuda a mitigar la tendencia de DT a sobreajustar los datos.
Cada árbol individual en el Bosque Aleatorio genera una predicción para la variable objetivo, pero no se espera que cada árbol sea altamente efectivo por sí solo. La fortaleza del método radica en la combinación de numerosos árboles, cada uno de los cuales opera en una región diferente del espacio de características. El Bosque Aleatorio se basa en una regla de decisión que cuenta los votos de cada árbol para determinar la predicción final. En teoría, un gran número de modelos relativamente no correlacionados que funcionan como un comité superarán a cualquier modelo individual.
Una ventaja fundamental del Bosque Aleatorio es que aprovecha la sensibilidad de DTs a los datos en los que se entrenan. Cada árbol individual se entrena con una muestra aleatoria del conjunto de datos, permitiendo la selección con reemplazo. Esto da como resultado árboles diferentes en el bosque, lo que mejora la generalización del modelo.
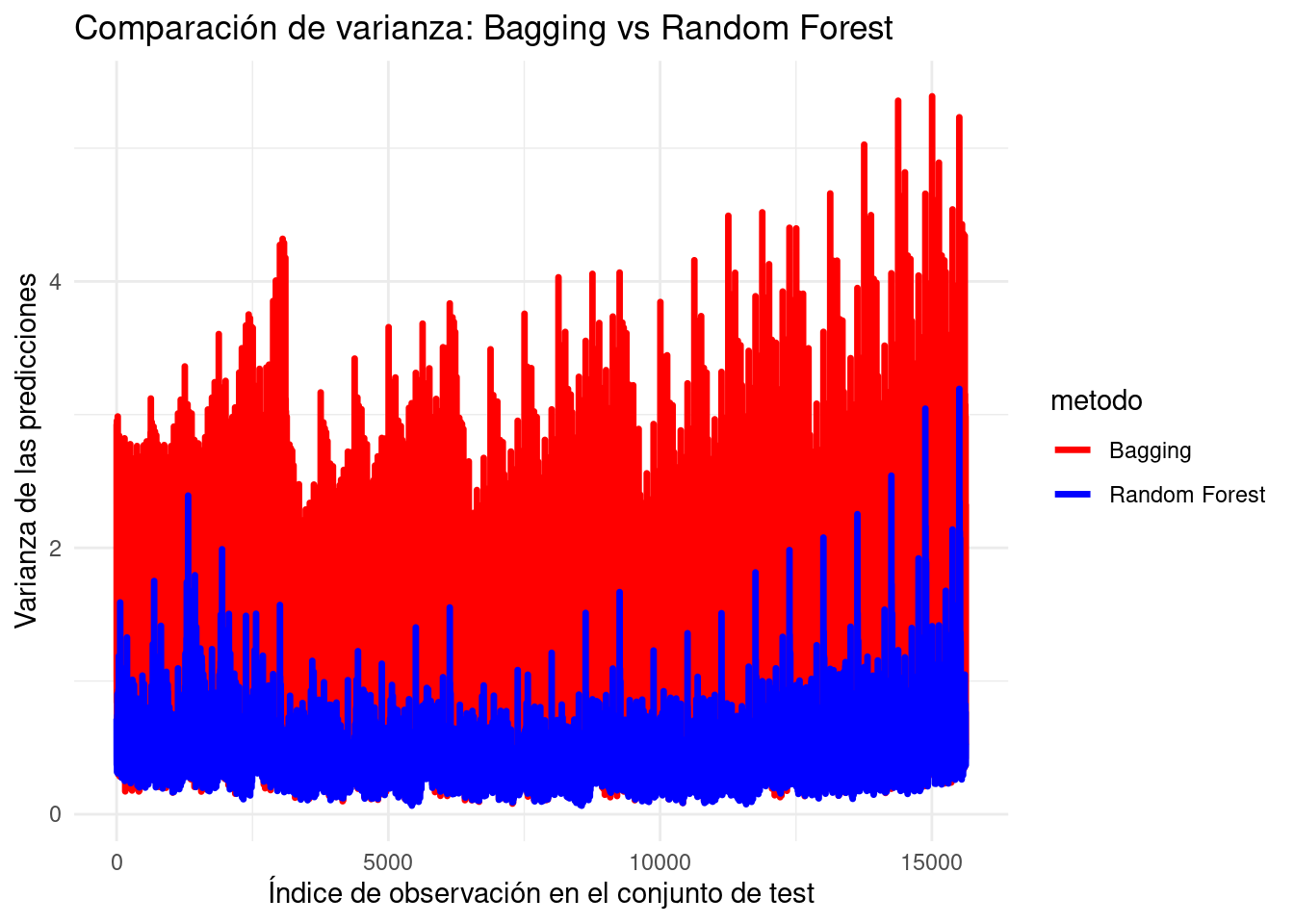
Varianza y correlación entre estimadores
Ya vimos que una de las fortalezas de Bagging es que, al promediar el resultado de distintos estimadores, se reduce la varianza inherente de los árboles de decisión. No obstante, dado que se entrenan \(M\) árboles en diferentes subconjuntos del conjunto de datos, los estimadores resultantes no son independientes, sino que están correlacionados. Esto es similar a obtener distintas muestras dependientes de la misma variable aleatoria: los estimadores son idénticamente distribuidos (i.d.), pero no independientes (i.i.d.).
La correlación entre estos estimadores tendrá un impacto en la reducción de varianza que se puede alcanzar con el promedio. Para mitigar esta correlación, Random Forest introduce una modificación clave: además de entrenar cada árbol con distintos subconjuntos del conjunto de datos (como en Bagging), también se entrena cada árbol con un subconjunto aleatorio de las variables predictoras \(\mathbf{x}_j\).
Desde un punto de vista téorico, consideremos \(M\) variables aleatorias \(\mathbf{X}_1,\mathbf{X}_2,\dots, \mathbf{X}_M\) que son idénticamente distribuidas con varianza \(\sigma^{2}\) pero no independientes, es decir, tienen correlación no nula. En particular,supongamos que la correlación entre cualquier par de ellas es \[Corr(\mathbf{X}_i,\mathbf{X}_j)=\rho, \ \ i\neq j.\]
La varianza del promedio de estos estimadores se obtiene como \[Var\left(\frac{1}{M} \sum_{i=1}^{M}\mathbf{X}_i \right)=\frac{\sum_{i=1}^{M} Var(\mathbf{X}_i) + 2\sum_{i< j}Cov(\mathbf{X}_i,\mathbf{X}_j)}{M^{2}}.\] Dado que \(Corr(\mathbf{X}_i,\mathbf{X}_j) = \rho = \frac{Cov(\mathbf{X}_i,\mathbf{X}_j)}{\sigma_{\mathbf{X}_i}\sigma_{\mathbf{X}_j}}\), tenemos que \(Cov(\mathbf{X}_i,\mathbf{X}_j)=\rho \sigma^{2}\). Además, como hay \[{M \choose 2} = \frac{M(M-1)}{2}\] pares de términos correlacionados, sustituimos en la ecuación anterior
\[Var\left(\frac{1}{M} \sum_{i=1}^{M}\mathbf{X}_i \right) = \frac{1}{M^{2}}\left(M \sigma^{2} + 2\frac{M(M-1)}{2}\rho \sigma^{2}\right)=\frac{M \sigma^{2} +M(M-1)\rho \sigma^{2}}{M^{2}}.\] Reordenando obtenemos \[Var\left(\frac{1}{M} \sum_{i=1}^{M}\mathbf{X}_i \right) = \rho \sigma^{2} + \frac{(1-\rho)}{M}\sigma^{2}.\] Aquí vemos que, a medida que a \(M \longrightarrow \infty\), el segundo término tiende a \(0\), pero el primer término permanece inalterado. Esto implica que la reducción de la varianza está limitada por la correlación entre los estimadores: si la correlación es alta (\(\rho\) cercano a 1), la varianza del promedio será elevada; si es baja (\(\rho\) cercano a 0), la reducción de varianza será mayor.
Random Forest introduce una estrategia para reducir esta correlación: seleccionar aleatoriamente un subconjunto de \(q \leq p\) variables predictoras para cada árbol, en lugar de usar todas las \(p\) variables disponibles.
En clasificación, se suele tomar \(q=\lfloor \sqrt{p} \rfloor\).
En regresión, se suele tomar \(q=\lfloor p/3 \rfloor\).
Al disminuir el número de variables disponibles para cada árbol, se fuerza a que los árboles tomen decisiones basadas en diferentes conjuntos de características, lo que aumenta la diversidad entre ellos y reduce la correlación entre los estimadores. Como resultado, la reducción de la varianza se vuelve más efectiva, mejorando la estabilidad y generalización del modelo.
Algoritmo
Así, el algoritmo del Random Forest es:
Inicialización. Definir los hiperparámetros del algoritmo: número de árboles \(M\) que compondrán el bosque, número de variables a considerar $q$, tamaño mínimo de los nodos terminales \(n_{nodo}\), etc.
Creación de cada árbol. Para cada árbol \(m\), \(m=1,\dots, M\):
Tomar una muestra bootstrap de tamaño \(n\) del conjunto de entrenamiento
Crear un árbol \(T_m\) usando la muestra bootstrap y en base a los siguientes pasos:
Selección de variables: Seleccionar \(q\) variables aleatoriamente de entre las \(p\) disponibles
Cálculo de la mejor división: Para las \(q\) variables seleccionadas, escoger la mejor variable y la mejor división (por ejemplo, minimizando la impureza de Gini o la entropía para clasificación, o el error cuadrático medio para regresión)
División del nodo: Dividir el nodo en 2 nodos hijos en función de la variable seleccionada y la mejor división
Recursión: Repetir el proceso de división recursivamente para cada nodo hijo hasta que se alcance un criterio de detención (máxima profundidad, número mínimo de muestras en un nodo, etc.).
Output y predicción. Devolver el ensamblado de árboles \(\{T_{m}\}_{m=1}^{M}\). Para hacer una predicción total, cada árbol realiza una predicción individual:
En clasificación: Utilizar el voto mayoritario entre las predicciones de todos los árboles.
En regresión: Promediar las predicciones de todos los árboles.
NotaSimulación para comparar la varianza de Bagging y Random Forest
library(randomForest)
randomForest 4.7-1.2
Type rfNews() to see new features/changes/bug fixes.
Attaching package: 'randomForest'
The following object is masked from 'package:dplyr':
combine
The following object is masked from 'package:ggplot2':
margin
library(ggplot2)# Generación de datos artificialesset.seed(42)n <-200x1 <-runif(n, -2, 2)x2 <-runif(n, -2, 2)x3 <-runif(n, -2, 2)x4 <-runif(n, -2, 2)x5 <-runif(n, -2, 2)x6 <-runif(n, -2, 2)y <- x1^3+ x2^2- x3 +0.5*x4 -0.7*x5 +0.3*x6 +rnorm(n, sd=1)data <-data.frame(x1, x2, x3, x4, x5, x6, y)# Número de repeticiones y datos testn_reps <-100x_test <-expand.grid(x1 =seq(-2, 2, length.out=5),x2 =seq(-2, 2, length.out=5),x3 =seq(-2, 2, length.out=5),x4 =seq(-2, 2, length.out=5),x5 =seq(-2, 2, length.out=5),x6 =seq(-2, 2, length.out=5))# Matrices para guardar resultadospred_bagging <-matrix(NA, n_reps, nrow(x_test))pred_rf <-matrix(NA, n_reps, nrow(x_test))for (i in1:n_reps) { sample_idx <-sample(1:n, n, replace =TRUE) train_data <- data[sample_idx, ]# Bagging (Random Forest sin selección de variables, mtry = número total de predictores) bagging_model <-randomForest(y ~ ., data=train_data, mtry=6) pred_bagging[i, ] <-predict(bagging_model, x_test)# Random Forest (mtry = sqrt(número total de predictores)) rf_model <-randomForest(y ~ ., data=train_data, mtry=round(sqrt(6))) pred_rf[i, ] <-predict(rf_model, x_test)}# Calcular la varianza de las predicciones en el conjunto de testvar_bagging <-apply(pred_bagging, 2, var)var_rf <-apply(pred_rf, 2, var)df_var <-data.frame(x_test =rep(1:nrow(x_test), 2),varianza =c(var_bagging, var_rf),metodo =rep(c("Bagging", "Random Forest"), each=nrow(x_test)))# Gráfico con ggplotggplot(df_var, aes(x = x_test, y = varianza, color = metodo)) +geom_line(linewidth =1.2) +labs(title ="Comparación de varianza: Bagging vs Random Forest",x ="Índice de observación en el conjunto de test",y ="Varianza de las predicciones") +theme_minimal() +scale_color_manual(values =c("red", "blue"))
Muestras Out-of-the-Bag (OOB)
En Random Forest, cada árbol se entrena utilizando una muestra aleatoria de los datos de entrenamiento mediante muestreo con reemplazo (bootstrap sampling). Esto significa que no todos los ejemplos del conjunto de datos original se utilizan en la creación de cada árbol. Las muestras OOB son aquellas observaciones que no se incluyen en el entrenamiento de un árbol específico, es decir, aquellas que no han sido seleccionadas en el muestreo de bootstrap para ese árbol en particular.
Una de las características importantes de las muestras OOB es que se pueden usar como un conjunto de validación para evaluar el rendimiento del modelo de manera eficiente y sin necesidad de particionar los datos. De esta forma, se obtiene una estimación del error del modelo similar a la validación cruzada (cross-validation), pero sin tener que realizar particionamiento explícito del conjunto de datos. Cada vez que se construye un árbol, se predice para las observaciones OOB de ese árbol, y luego se calcula el error OOB promedio, el cual puede utilizarse para obtener una estimación del rendimiento general del modelo. Esto proporciona una forma de evaluar el modelo sin necesidad de usar un conjunto de datos adicional para validación, como en la validación cruzada tradicional.
Este método de evaluación es especialmente útil cuando el número de datos es limitado y permite aprovechar todo el conjunto de entrenamiento de manera más eficiente.
Importancia de las variables
Uno de los beneficios de Random Forest es que permite evaluar la importancia de cada variable en la predicción del modelo. Existen dos enfoques principales para medir esta importancia:
Importancia basada en la impureza (Gini o entropía). Durante la construcción de cada árbol en el bosque, se seleccionan variables en los nodos de decisión en función de su capacidad para reducir la impureza en la clasificación (medida por el índice de Gini o la entropía) o el error cuadrático medio en regresión. La importancia de una variable se calcula como la reducción total de impureza que causa a lo largo de todos los nodos en los que es seleccionada, promediada sobre todos los árboles del bosque. En términos generales, si una variable aparece con frecuencia en los nodos de división y contribuye notablemente a reducir la impureza, se considera más importante.
Una desventaja de este método es que tiende a sobrestimar la importancia de las variables con más valores posibles (variables continuas frente a categóricas con pocas categorías).
Importancia basada en permutaciones. En este enfoque, se mide la importancia de una variable observando cómo cambia la precisión del modelo cuando se permutan aleatoriamente los valores de dicha variable en el conjunto de prueba o en las muestras OOB (Out-of-Bag). Si una variable es importante para el modelo, al permutar sus valores y hacer que se pierda la relación existente entre la variable y el target se espera que el rendimiento del modelo disminuya. La importancia de la variable se mide como la diferencia en rendimiento antes y después de la permutación, promediada sobre todos los árboles.
La ventaja de este método es que es más confiable porque evalúa la importancia en términos de la contribución real de la variable a la predicción del modelo y no se ve afectado por el número de categorías o valores posibles. El inconveniente es que es más costoso computacionalmente, ya que requiere realizar múltiples predicciones con los valores permutados.
En la práctica, se recomienda analizar ambos métodos para obtener una visión más completa del impacto de cada variable en el modelo.
Que el Random Forest proporcione información sobre la importancia de las variables resulta muy útil e interesante puesto que, al combinar gran cantidad de árboles de decisión, se pierde la interpretabilidad que caracteriza a dicho modelo.
Aplicamos este nuevo método de ML a nuestro conjunto de datos de bank.
Call:
randomForest(formula = as.factor(deposit) ~ ., data = df, importance = TRUE, proximity = TRUE)
Type of random forest: classification
Number of trees: 500
No. of variables tried at each split: 2
OOB estimate of error rate: 19.33%
Confusion matrix:
no yes class.error
no 2343 565 0.1942916
yes 514 2159 0.1922933
plot(rf)
# sobre la partición de pruebadf.test <- bank.test %>%mutate(fmarital=as.factor(marital))%>% dplyr::select(age,job,housing,marital=fmarital,education,duration,poutcome,balance,deposit)df.test$deposit=as.factor(df.test$deposit)prediction.rf <-predict(rf, df.test,type="prob")[,2]clase.pred.rf=ifelse(prediction.rf>0.5,"yes","no")cf <-confusionMatrix(as.factor(clase.pred.rf), as.factor(df.test$deposit),positive="yes")print(cf)
Confusion Matrix and Statistics
Reference
Prediction no yes
no 1155 248
yes 313 1074
Accuracy : 0.7989
95% CI : (0.7836, 0.8137)
No Information Rate : 0.5262
P-Value [Acc > NIR] : < 2.2e-16
Kappa : 0.5977
Mcnemar's Test P-Value : 0.006891
Sensitivity : 0.8124
Specificity : 0.7868
Pos Pred Value : 0.7743
Neg Pred Value : 0.8232
Prevalence : 0.4738
Detection Rate : 0.3849
Detection Prevalence : 0.4971
Balanced Accuracy : 0.7996
'Positive' Class : yes
Podemos averiguar la importancia de las variables en el modelo.
Las tres primeras columnas hacen referencia a la medida MeanDecreaseAccuracy. La disminución del accuracy se mide permutando los valores del target en las muestras OOB y calculando la disminución correspondiente. Esto se hace para cada árbol y con sus muestras OOB correspondientes para obtener la media y la desviación estándar. La segunda medida es la disminución total de impurezas de nodos a partir de la división en la variable, promediada en todos los árboles.
varImpPlot(rf)
Este gráfico muestra la importancia de las variables, ordenadas de mayor a menor. En este caso particular la variable más relevante dentro del modelo es la duración de la llamada.
Además podemos obtener una visualización del funcionamiento del Bosque con la función MDSplot:
#MDSplot(rf,as.factor(df$deposit),k=3)
La función grafica las \(k\) primeras componentes principales (ver Capítulo 4) de la matriz de proximidad, que es una matriz de medidas de proximidad entre la observaciones de entrenamiento (basada en la frecuencia con que los pares de puntos de datos se encuentran en los mismos nodos terminales).
7.6.2 Boosting
El ensamblado de Boosting es una técnica de ML que se utiliza para mejorar el rendimiento de los modelos de clasificación o regresión.A diferencia del Bagging, que entrena múltiples modelos en paralelo y los promedia, el Boosting entrena modelos secuencialmente, dando mayor peso a las observaciones mal clasificadas en cada iteración.
El proceso de Boosting funciona de la siguiente manera:
Comienza con un modelo base (generalmente un modelo simple o débil), que se entrena en el conjunto de datos original.
Luego, se evalúa el rendimiento del modelo base. Las instancias clasificadas incorrectamente o las predicciones con errores se ponderan más fuertemente para la siguiente iteración.
En la siguiente iteración, se entrena un segundo modelo débil, pero esta vez se da más énfasis a las instancias que se clasificaron incorrectamente en la iteración anterior.
Los resultados de los modelos base se combinan ponderando sus predicciones en función de su rendimiento relativo. Los modelos que tienen un mejor rendimiento reciben más peso en la predicción final.
Este proceso de entrenamiento, evaluación y asignación de pesos se repite durante varias iteraciones (también llamadas etapas de Boosting). Cada nuevo modelo se enfoca en corregir las deficiencias del modelo anterior.
Al final de las iteraciones, se obtiene un modelo ensamblado fuerte que es capaz de mejorar significativamente la precisión de las predicciones en comparación con un solo modelo base.
Algunos algoritmos de Boosting populares incluyen AdaBoost, Gradient Boosting, y XGBoost.
NotaVentajas
Mejora del rendimiento: El Boosting es conocido por aumentar significativamente la precisión de las predicciones en comparación con modelos individuales o modelos base.
Reducción del sesgo: A través del proceso iterativo, el Boosting puede reducir el sesgo del modelo, lo que significa que se vuelve más capaz de ajustarse a los datos y hacer predicciones precisas.
Manejo de datos desequilibrados: Es eficaz en la clasificación de conjuntos de datos con clases desequilibradas, ya que se enfoca en las instancias mal clasificadas.
Adaptabilidad: Funciona bien con una variedad de algoritmos base, lo que brinda flexibilidad al elegir el modelo débil más adecuado.
Versatilidad: El Boosting se utiliza en problemas de clasificación y regresión, lo que lo hace adecuado para una amplia gama de aplicaciones.
Capacidad para detectar patrones complejos: Al mejorar iterativamente el modelo, el Boosting puede capturar patrones complejos en los datos y generar modelos más precisos.
Robustez: Puede manejar ruido en los datos y es menos propenso al sobreajuste en comparación con algunos otros métodos de aprendizaje automático.
NotaDesventajas
Sensibilidad al ruido: Los modelos de Boosting pueden ser sensibles al ruido y a valores atípicos en los datos. Si el conjunto de datos contiene observaciones erróneas o extremas, el Boosting puede sobreajustarse a estos valores inusuales.
Tiempo de entrenamiento: El proceso de Boosting implica entrenar múltiples modelos base de manera secuencial, lo que puede llevar más tiempo en comparación con otros métodos de ensamblado más simples.
Menos interpretabilidad: A medida que se agregan más modelos al ensamblado, la interpretabilidad del modelo general puede disminuir. Esto hace que sea más difícil comprender y explicar las predicciones del modelo.
Necesidad de ajuste de hiperparámetros: Los modelos de Boosting tienen varios hiperparámetros que deben ajustarse adecuadamente para obtener el mejor rendimiento. Encontrar la combinación óptima de hiperparámetros puede requerir tiempo y recursos computacionales.
Potencial sobreajuste: Si no se ajustan cuidadosamente los hiperparámetros, los modelos de Boosting pueden estar sujetos al sobreajuste, especialmente si el número de iteraciones (número de modelos base) es demasiado alto.
Requisitos de recursos computacionales: Dependiendo de la implementación y la cantidad de modelos base, los modelos de Boosting pueden requerir recursos computacionales significativos, lo que podría ser un problema en sistemas con recursos limitados.
7.6.2.1 AdaBoost: Algoritmo de Boosting Adaptativo
Uno de los algoritmos más representativos de Boosting es AdaBoost, que ajusta secuencialmente clasificadores débiles, aumentando el peso de las observaciones mal clasificadas en cada iteración.
Algoritmo AdaBoost
Inicializamos los pesos de las observaciones de la siguiente manera: \[w_i=1/n, i=1,\dots,n\] donde \(n\) es el número total de observaciones.
Bucle para \(m=1,\dots, M\). Para cada iteración \(m\) en el algoritmo AdaBoost, realizamos los siguientes pasos:
Ajustar el clasificador \(f_m(\mathbf{x})\). Ajustamos un clasificador \(f_m(\mathbf{x})\) al conjunto de entrenamiento usando los pesos \(w_i\). Este clasificador \(f_m(\mathbf{x},\phi_m)\) es un modelo base que depende de los hiperparámetros \(\phi_m\), como un árbol de decisión.
Calcular el error ponderado. El error ponderado de \(f_m(\mathbf{x})\) se calcula como \[error_{m}=\frac{\sum_{i=1}^{n}w_i I(y_i \neq f_{m}(\mathbf{x}_i))}{\sum_{i=1}^{n}w_i}\] donde \(I(y_i \neq f_{m}(\mathbf{x}_i))\) es la función indicadora que toma el valor 1 si la predicción \(f_m(\mathbf{x})\) es incorrecta, y 0 en caso contrario. Este error indica lo bien o mal que está funcionando el clasificador.
Calcular el coeficiente \(\alpha_m\). El coeficiente \(\alpha_m\), que que ponderará la contribución del clasificador \(f_m(\mathbf{x})\) en la predicción final, se calcula con la siguiente fórmula: \[\alpha_m = log\left(\frac{1-error_m}{error_m} \right)\] Este coeficiente mide la “importancia” del clasificador en la actualización del modelo acumulado.
Actualizar los pesos de las observaciones Los pesos de las observaciones se actualizan de acuerdo con su desempeño en la clasificación. Las observaciones mal clasificadas recibirán un aumento en el peso, lo que hace que el siguiente clasificador se enfoque más en ellas. Los nuevos pesos \(w_i^{(m+1)}\) se calculan como: \[w_i^{(m+1)} =w_i^{(m)} exp(\alpha_m I(y_i \neq f_{m}(\mathbf{x}_i))), i=1\dots, n\] donde \(y_i\) es la etiqueta real de la observación \(i\). El término \(exp(\alpha_m I(y_i \neq f_{m}(\mathbf{x}_i)))\) aumenta el peso de las observaciones mal clasificadas.
El modelo final \(F(\mathbf{x})\) es la combinación de los clasificadores base ajustados en cada iteración, ponderados por los coeficientes \(\alpha_{m}\). La predicción final se calcula como: \[F(\mathbf{x})=sign\left( \sum_{m=1}^{M} \alpha_{m} f_{m}(\mathbf{x})\right)\] Este modelo es una suma ponderada de los clasificadores base, donde los clasificadores más precisos (con valores más altos de \(\alpha_{m}\)) tendrán más influencia en la predicción final.
Relación con modelo aditivo
En AdaBoost, la expresión de la función de predicción es: \(F(\mathbf{x})=sign\left( \sum_{m=1}^{M} \alpha_{m} f_{m}(\mathbf{x})\right)\), es decir, lo que estamos haciendo es construir un modelo aditivo. De manera más general, podemos expresar este tipo de modelos como: \[F(\mathbf{x})=\sum_{m=1}^{M} \alpha_m f_m(\mathbf{x},\phi_m)\]
donde \(\alpha_m\) son coeficientes que ponderan la contribución de cada componente, y \(f_m(\mathbf{x};\phi_m)\) son las funciones base individuales parametrizadas por \(\phi_m\) que se ajustan secuencialmente a los datos.
Este esquema de ajuste secuencial aparece en otros modelos de Aprendizaje Automático como splines o redes neuronales. Sin embargo, en AdaBoost, la diferencia clave está en la función de pérdida utilizada y en la manera en que los modelos base van corrigiendo errores.
Ajuste secuencial en modelos aditivos
Dado que la optimización conjunta de todos los parámetros/hiperparámetros en \(F(\mathbf{x})\) de forma global es compleja, se adopta un procedimiento secuencial: en cada iteración se ajusta un nuevo modelo \(f_m(\mathbf{x};\phi_m)\) para minimizar el error con respecto al modelo acumulado hasta el momento \(F_{m-1}(\mathbf{x})\).
Esto se hace mediante la optimización de la función de pérdida en cada paso, sin tener que considerar el conjunto completo de modelos de una vez. Esto se traduce en la siguiente optimización
\[(\alpha_m,\phi_m)= arg min_{\alpha, \phi} \sum_{i=1}^{n}L(y_i, F_{m-1}(\mathbf{x}_i)+ \alpha f(\mathbf{x}_i,\phi))\] y la función se actualiza como: \[F_{m}(\mathbf{x})=F_{m-1}(\mathbf{x}) + \alpha_m f(\mathbf{x},\phi_m).\] En \(m=0\) se proporciona un ajuste inicial que puede ser, por ejemplo, la media en el caso de regresión \(f_0(\mathbf{x})=\overline{y}\) o las proporciones a priori en el caso de clasificación.
Si utilizamos la función de pérdida cuadrática \(L(y,F(\mathbf{x}))=(y-F(\mathbf{x}))^{2}\), obtenemos la siguiente expresión para cada iteración: \[
L(y_i,F(\mathbf{x}_i))=L(y_i,F_{m-1}(\mathbf{x}_i)+\alpha f(\mathbf{x}_i,\phi))=(\underbrace{y_i-(F_{m-1}(\mathbf{x}_i)}_{r_{i}^{(m)}}+\alpha f(\mathbf{x}_i,\phi)))^{2} = (r_{i}^{(m)}-\alpha f(\mathbf{x}_i,\phi))^{2}\]
donde \(r_{i}^{(m)}\) representa los residuos del modelo actual (\(F_{m-1}(\mathbf{x}_i)\)). ¿Cómo se interpreta esto? La interpretación de esta ecuación es que, en cada paso, el nuevo término \(\alpha f(\mathbf{x}_i,\phi)\) busca ajustar los errores que ha cometido el modelo hasta el momento.
Caso de AdaBoost: Función de pérdida exponencial En AdaBoost, en lugar de la pérdida cuadrática, se emplea una función de pérdida exponencial: \(L(y,f(\mathbf{x}))=exp(-yf(\mathbf{x})).\) Es decir, la función de pérdida que se minimiza es: \[L(y_i,F_{m-1}(\mathbf{x}_i)+\alpha f_m(\mathbf{x},\phi_m))=exp(-y_i(F_{m-1}(\mathbf{x}_i) + \alpha f_m(\mathbf{x},\phi_m)))\] donde \(y_i\) es la etiqueta de la observación \(i\), \(F_{m-1}(\mathbf{x}_i)\) es la predicción acumulada hasta la iteración \(m-1\), y \(\alpha_m\) es el coeficiente que ajusta la contribución de \(f_m(\mathbf{x},\phi_m).\)
Siguiendo la misma lógica del ajuste secuencial, el nuevo modelo se obtiene minimizando:
Una forma de reformular esta expresión es definiendo los pesos \(w_i^{(m)}\) de cada observación en la iteración \(m\): \(w_i^{(m)}=exp(-y_i F_{m-1}(\mathbf{x}_i))\) puesto que \(w_i^{(m)}\) es independiente tanto de \(\alpha\) como de \(f_m\). Este término refleja la importancia relativa de cada instancia en la siguiente iteración: los errores pasados aumentan su influencia y afectan más al siguiente modelo. Este término refleja la importancia relativa de cada instancia en la siguiente iteración: los errores pasados aumentan su influencia y afectan más al siguiente modelo (el objetivo es reducir progresivamente el error al hacer que el nuevo modelo ajuste los fallos del conjunto de modelos acumulado).
A partir de aquí, minimizando la función de pérdida exponencial (derivando \(L\) con respecto a \(\alpha_m\) e igualando a 0), se obtiene la expresión del coeficiente \(\alpha_m\):
\[\alpha_m = \frac{1}{2}\ln\left(\frac{1-error_m}{error_m}\right)\] donde el error de clasificación en la iteración \(m\) está dado por:
\[error_m = \frac{\sum_{i=1}^{n} w_i^{(m)}I(y_i \neq f_{m}(\mathbf{x}_i))}{\sum_{i=1}^{n} w_i^{(m)}}.\] Finalmente, los pesos se actualizan según \(w_i^{(m+1)}=w_i^{(m)} exp(-y_i \alpha_m f_{m}(\mathbf{x}_i)).\) Usando que \(-y_i f_m(\mathbf{x}_i) = 2 I(y_i \neq f_m(\mathbf{x}_i))-1\), podemos escribir la actualización de pesos como \[w_i^{(m+1)} = w_i^{(m)} e^{\alpha_m I(y_i \neq f_m(\mathbf{x}_i))} e^{-\alpha_m}.\] El término \(e^{-\alpha_m}\) es constante y no afecta la normalización relativa de los pesos, por lo que puede ignorarse. Con esto, habríamos obtenido todos los valores del algoritmo Adaboost.
En resumen:
AdaBoost sigue el mismo principio de ajuste secuencial que los modelos aditivos.
La diferencia clave es el uso de una función de pérdida exponencial, que refuerza la influencia de los ejemplos mal clasificados.
En cada iteración, AdaBoost ajusta un nuevo modelo para corregir los errores acumulados, asignando mayores pesos a los ejemplos que han sido más difíciles de clasificar.
La actualización de los pesos garantiza que el ensamble final esté compuesto por modelos base especializados en corregir errores previos.
7.6.2.2 Gradient Boosting
El algoritmo de Gradient Boosting es un método de aprendizaje supervisado que construye un modelo de predicción de manera secuencial, ajustando cada modelo subsecuente para corregir los errores del modelo acumulado. A diferencia de AdaBoost, que utiliza una función de pérdida exponencial, Gradient Boosting puede usar cualquier función de pérdida y optimizarla con el algoritmo de descenso de gradiente.
Relación con el descenso de gradiente
La clave de Gradient Boosting es que, en cada paso, el modelo nuevo se ajusta para minimizar la función de pérdida tomando en cuenta el gradiente de la pérdida con respecto a las predicciones actuales del modelo.
De manera más formal, para un conjunto de datos \(\{\mathbf{x}_i,y_i\}_{i=1}^{n}\), la función de pérdida generalizada es: \[L(y_i,F(\mathbf{x}_i)).\] El objetivo es minimizar la pérdida total: \[\sum_{i=1}^{n} L(y_i,F(\mathbf{x}_i)).\] En cada iteración \(m\), el modelo ajusta una nueva función \(f_m(\mathbf{x})\) que minimiza la pérdida respecto al modelo acumulado \(F_{m-1}(\mathbf{x}).\) La optimización se realiza usando el gradiente de la función de pérdida.
Paso de Gracient Boosting
1. Inicialización: Se inicia con una predicción constante \(F_0(\mathbf{x})\), que generalmente es la media o mediana de las respuestas \(y_i\) en el caso de regresión.
2. Iteración: Para cada iteración \(m=1,2\dots,M\):
Se calcula el residuo o el gradiente de la función de pérdida con respecto al modelo acumulado \(F_{m-1}(\mathbf{x})\). Este residuo indica cómo cambiar el modelo para reducir la pérdida: \[r_{i}^{(m)}=-\frac{\partial L(y_i,F_{m-1}(\mathbf{x}_i))}{\partial F_{m-1}(\mathbf{x}_i)}\]
Se ajusta un modelo \(f_m(\mathbf{x})\) para predecir estos residuos. Este modelo es generalmente un árbol de decisión pequeño (árbol débil).
Se optimiza el hiperparámetro \(\alpha_m\) que, junto a la tasa de aprendizaje \(\eta\) controla la magnitud de la corrección del modelo en cada paso: \[\alpha_m = arg min_{\alpha} \sum_{i=1}^{n} L(y_i,F_{m-1}(\mathbf{x}_i+\alpha f_{m}(\mathbf{x}_i)))\]
Finalmente, se actualiza el modelo acumulado: \[F_m(\mathbf{x})=F_{m-1}(\mathbf{x}) + \eta \alpha_m f_{m}(\mathbf{x})\]
Resultado: El modelo final es: \[F(\mathbf{x}) = F_0(\mathbf{x}) + \eta \sum_{m=1}^{M} \alpha_m f_m(\mathbf{x})\]
¿Cómo se calculan los\(\alpha_m\)?
El coeficiente \(\alpha_m\) controla cuánto contribuye cada modelo \(f_m(\mathbf{x})\) al modelo acumulado (determina el tamaño del paso para la actualización). Se determina minimizando la función de pérdida total: \[\alpha_m = \arg\min_{\alpha}\sum_{i=1}^{n} L(y_i,F_{m-1}(\mathbf{x}_i)+\alpha f_{m}(\mathbf{x}_i))\]
Así, los \(\alpha_m\) se obtienen mediante un proceso de optimización. Si la pérdida es diferenciable, se calcula la derivada de la función de pérdida con respecto a \(\alpha\) y obtenemos la magnitud en la que debemos actualizar el modelo.
Tasa de aprendizaje
La tasa de aprendizaje \(\eta\) regula también la magnitud de las actualizaciones en cada iteración de Gradient Boosting. En lugar de actualizar el modelo acumulado sólo con \(\alpha_m\), se escala su impacto mediante \(\eta\):
\[F_{m}(\mathbf{x})=F_{m-1}(\mathbf{x})+\eta \alpha_{m} f_{m}(\mathbf{x})\] La tasa de aprendizaje \(\eta\) es crucial para el comportamiento general del modelo de Gradient Boosting, asegurando que las actualizaciones sean de la magnitud adecuada para lograr que haya mejora pero no se sobreajuste. Es decir, controla el balance entre velocidad de convergencia y sobreajuste.
NotaCaso de regresión con pérdida cuadrática
En Gradient Boosting, la idea clave es que en cada iteración se busca minimizar una función de pérdida ajustando un modelo a la dirección del descenso del gradiente de dicha función.
Si tomamos la pérdida cuadrática \[L(y_i,F(\mathbf{x}_i))=(y_i - F(\mathbf{x}_i))^{2},\] el gradiente de la pérdida respecto a \(F_{m-1}(\mathbf{x}_i)\) es \[\frac{\partial L(y_i,F_{m-1}(\mathbf{x}_i))}{\partial F_{m-1}(\mathbf{x}_i)}=-2(y_i-F_{m-1}(\mathbf{x}_i)).\] El método de descenso del gradiente indica que, para minimizar la función de pérdida, hay que moverse en la dirección opuesta a su gradiente. En este caso, los residuos \[r_i^{(m)}=-\frac{\partial L(y_i,F_{m-1}(\mathbf{x}_i))}{\partial F_{m-1}(\mathbf{x}_i)}=2(y_i-F_{m-1}(\mathbf{x}_i))\] nos dan esa dirección, lo que significa que el siguiente modelo \(f_{m}(\mathbf{x})\) se ajusta para predecir los residuos \(r_i^{(m)}\), corrigiendo los errores del modelo anterior.
Una vez ajustado \(f_{m}(\mathbf{x})\), se determina el coeficiente \(\alpha_{m}\) minimizando
La función de pérdida logarítmica (log-loss) se basa en la idea de maximizar la verosimilitud de los datos bajo un modelo probabilístico.
En un problema de clasificación binaria, donde \(y_i \in \{0,1\}\), el objetivo es construir un modelo que prediga la probabilidad de que una observación pertenezca a la clase \(y_i=1\), dado un conjunto de características \(\mathbf{x}_i\).
Dado un conjunto de datos con \(n\) observaciones, asumimos que cada \(y_i\) sigue una distribución de Bernoulli con probabilidad \(p_i\): \[P(y_i | \mathbf{x}_i) = p_i^{y_i}(1-p_i)^{1-y_i}.\] Como has estudiado en asignaturas previas del Grado en Ciencia e Ingeniería de Datos, dado un conjunto de datos \(\{(\mathbf{x}_i,y_i)\}_{i=1}^{n}\), la verosimilitud conjunta es el producto de todas las probabilidades individuales: \[L(y,\mathbf{x})=\prod_{i=1}^{n} p_i^{y_i}(1-p_i)^{1-y_i}.\] Para facilitar los cálculos, tomamos el logaritmo de esta verosimilitud: \[\log L(y,\mathbf{x})=\sum_{i=1}^{n} \left[ y_i \log p_i + (1-y_i) \log (1-p_i) \right].\] La función de pérdida logarítmica es la versión negativa de este logaritmo de la verosimilitud: \[L(y_i,F(\mathbf{x}_i))=- \left[ y_i \log p_i + (1-y_i) \log (1-p_i) \right].\] Esta función mide qué cómo se ajustan las probabilidades predichas \(p_i\) a las etiquetas reales \(y_i\). El objetivo al entrenar un modelo de clasificación es minimizar esta función de pérdida para mejorar la calidad de las predicciones.
Con el siguiente código puedes ver mejor cómo funciona la pérdida log-loss.
library(ggplot2)# Función de pérdida log-losslog_loss <-function(y, p) {- (y *log(p) + (1- y) *log(1- p))}# Rango de probabilidadesp_valores <-seq(0.01, 0.99, by =0.01)loss_y1 <-log_loss(1, p_valores) # Cuando la etiqueta real es 1loss_y0 <-log_loss(0, p_valores) # Cuando la etiqueta real es 0loss_df <-data.frame(p =rep(p_valores, 2),loss =c(loss_y1, loss_y0),y =rep(c(1, 0), each =length(p_valores)))# Gráficoggplot(loss_df, aes(x = p, y = loss, color =factor(y))) +geom_line(linewidth =1.2) +labs(title ="Función de pérdida Log-Loss",x ="Probabilidad predicha (p)",y ="Log-Loss",color ="Etiqueta real") +theme_minimal() +theme(legend.position ="top")
NotaCaso de clasificación con pérdida log-loss
En clasificación, la función de pérdida más utilizada en Gradient Boosting es la pérdida logarítmica (log-loss) para problemas de clasificación binaria. A diferencia de la regresión, donde predecimos valores continuos, en clasificación binaria nuestro objetivo es predecir la probabilidad de que una observación pertenezca a una de las dos clases.
En lugar de predecir directamente la etiqueta \(y_i\), el modelo ajusta una función \(F(\mathbf{x}_i)\) para modelar la probabilidad de pertenecer a la clase \(y_i=1\). Esta probabilidad se calcula mediante la siguiente fórmula logística: \[p_i = P(y_i=1 | \mathbf{x}_i)=\frac{1}{1+e^{-F(\mathbf{x}_i)}}.\] Esta transformación asegura que las predicciones \(p_i\) se encuentren siempre en el intervalo \([0,1]\), que es lo necesario para una probabilidad.
La función de pérdida logarítmica es \[L(y_i,F(\mathbf{x}_i))=-y_i \log p_i - (1-y_i)\log(1-p_i).\] El gradiente de la función de pérdida respecto a la predicción \(F_{m-1}(\mathbf{x}_i)\) del modelo en la iteración \(m-1\) es \[\frac{\partial L(y_i,F_{m-1}(\mathbf{x}_i))}{\partial F_{m-1}(\mathbf{x}_i))}=p_i - y_i.\] Este gradiente muestra cómo cambia la función de pérdida a medida que ajustamos \(F(\mathbf{x}_i)\). Como el gradiente está dado por la diferencia entre la probabilidad predicha y la verdadera clase, podemos interpretarlo como una medida de error en la predicción de la probabilidad.
Por tanto, los pseudo-redisuos son \(r_i^{(m)}=-(p_i - y_i),\) que representan cuánto se equivoca la predicción de la probabilidad respecto a la verdadera clase \(y_i\).
Al igual que en regresión, en cada iteración se ajusta un modelo \(f_{m}(\mathbf{x}_i)\) para predecir estos errores \(r_i^{(m)}\) cometidos en iteraciones previas.
PrecauciónTarea
Cada función de pérdida tiene sus ventajas y desventajas, y la elección de la función adecuada puede tener un impacto significativo en el rendimiento del modelo. Existen diversas funciones de pérdida tanto para regresión como para clasificación. Te animamos a hacer una búsqueda en la literatura.
7.6.2.3 XGBoost
XGBoost, que significa “Extreme Gradient Boosting”, es un modelo de ML de tipo ensamblado que ha ganado gran popularidad en competiciones de ciencia de datos y aplicaciones prácticas debido a su alto rendimiento y eficacia en una variedad de problemas de clasificación y regresión. XGBoost es una poderosa técnica de ML que ha demostrado su eficacia en una amplia gama de aplicaciones. Su capacidad para manejar datos ruidosos, seleccionar características importantes y su velocidad de entrenamiento lo hacen valioso tanto para profesionales de datos como para científicos de datos en competiciones y proyectos del mundo real.
XGBoost es una extensión del método de Gradient Boosting, que se centra en mejorar las debilidades de dicho algoritmo. ¿Cómo funciona el método del Gradient Boosting?. Lo vemos en la siguiente figura:
Como hemos dicho, XGBoost es una optimización del algoritmo de Gradient Boosting que mantiene su filosofía básica pero introduce mejoras clave para hacerlo más eficiente y robusto. Al igual que Gradient Boosting, XGBoost construye modelos de manera secuencial, ajustando nuevos árboles a los residuos de los anteriores, pero incorpora innovaciones que lo hacen superior en términos de velocidad, capacidad de generalización y escalabilidad:
Mejora computacional. Uno de los mayores avances de XGBoost es su eficiencia computacional. XGBoost optimiza su ejecución mediante:
Cálculo en bloques: Utiliza estructuras de datos optimizadas para mejorar el acceso a la memoria y reducir el tiempo de cómputo.
Paralelización: Aprovecha el procesamiento paralelo en la construcción de los árboles, dividiendo los datos de manera más eficiente.
Regularización para evitar sobreajuste. Incorpora términos de regularización en su función de pérdida, lo que mejora su capacidad de generalización. La función de pérdida que minimiza incluye un término adicional: \[L=\sum_{i=1}^{n} l(y_i,\hat{y}_i)+ \sum_{m=1}^{M} \Omega(f_m)\] donde \(\Omega(f_m)\) es un término de regularización que penaliza la complejidad de los árboles. Este término se define como: \[\Omega(f_m) = \gamma T + \frac{1}{2}\lambda \sum w^{2}\] donde \(T\) es el número de hojas del árbol (penaliza árboles grandes), \(w\) representa los pesos de las hojas (penaliza valores extremos de los nodos), \(\gamma\) y \(\lambda\) son hiperparámetros que controlan la regularización. Esta regularización hace que los árboles sean más simples y evita que el modelo se ajuste demasiado a los datos de entrenamiento.
Manejo eficiente de datos. XGBoost por ejemplo usa histogramas de búsqueda para reducir el número de comparaciones necesarias al dividir nodos.
Convergencia acelerada. También usa la segunda derivada de la función de pérdida para realizar actualizaciones más precisas en los pesos de los árboles. Esto equivale a usar una aproximación de Newton-Raphson en lugar de un simple descenso del gradiente, lo que acelera la convergencia del modelo.
##### xgb.Booster
call:
xgb.train(data = dtrain, nrounds = 10, objective = "binary:logistic",
max_depth = 2, eta = 0.3, nthread = 2)
# of features: 24
# of rounds: 10
Ahora podemos examinar e interpretar nuestro modelo XGBoost. Una forma en que podemos examinar nuestro modelo es observando una representación de la combinación de todos los DTs en nuestro modelo. Dado que todos los árboles tienen la misma profundidad podemos apilarlos unos encima de otros y elegir las características que aparecen con más frecuencia en cada nodo.
library(DiagrammeR)# Visualización del conjunto de árboles como una única unidad xgb.plot.multi.trees(model = modelo_01)
En esta figura, la parte superior del árbol está a la izquierda y la parte inferior a la derecha. En el caso de las características, el número que aparece al lado es la “calidad”, que ayuda a indicar la importancia de la característica en todos los árboles. Una mayor calidad significa que la característica es más importante. Por tanto, podemos decir que duration es con diferencia la característica más importante en todos nuestros árboles, tanto porque está más arriba en el árbol como porque su puntuación de calidad es muy alta.
Para los nodos “hoja”, el número es el valor medio del modelo devuelto a través de todos los árboles cuando una determinada observación terminó en esa hoja. Como estamos utilizando un modelo logístico, nos devuelve diciendo que el logaritmo de probabilidades en lugar de la probabilidad. Sin embargo, podemos convertir fácilmente las probabilidades logarítmicas en probabilidad.
# convertimos log odds a probabilidadesodds_to_probs <-function(odds){return(exp(odds)/ (1+exp(odds)))}# ejemplo odds_to_probs(-1.2496)
[1] 0.2227694
Así, en los árboles en los que una observación acabó en esa hoja, la probabilidad media de que una observación fuera un “yes” en la variable deposit es del \(22\%\).
¿Y si queremos ver rápidamente qué características son las más importantes? Podemos hacerlo creando, y luego trazando, la matriz de importancia, como sigue.
# importancia sobre cada característicaimportance_matrix <-xgb.importance(model = modelo_01)xgb.plot.importance(importance_matrix)
Pasamos a obtener las predicciones. El resultado es un vector de valores numéricos, cada uno representando la probabilidad de que un caso en particular pertenezca al valor \(1\) de nuestra variable objetivo. Es decir, la probabilidad de que esa observación sea un “yes”.
# Predicciones en la muestra de prueba# Predicciones (probabilidades)pred_prob <-predict(modelo_01, dtest)pred_class <- pred_prob >0.5# Tabla + matriz de confusióncbind(pred_class, df.test$deposit) %>%data.frame() %>%table() %>% caret::confusionMatrix(positive ="1")
Confusion Matrix and Statistics
V2
pred_class 0 1
0 1216 344
1 252 978
Accuracy : 0.7864
95% CI : (0.7707, 0.8015)
No Information Rate : 0.5262
P-Value [Acc > NIR] : < 2.2e-16
Kappa : 0.5701
Mcnemar's Test P-Value : 0.0001934
Sensitivity : 0.7398
Specificity : 0.8283
Pos Pred Value : 0.7951
Neg Pred Value : 0.7795
Prevalence : 0.4738
Detection Rate : 0.3505
Detection Prevalence : 0.4409
Balanced Accuracy : 0.7841
'Positive' Class : 1
ImportanteTime consuming
La tarea que más tiempo consume al usar el modelo XGBoost es encontrar los mejores hiperparámetros para alcanzar la mayor precisión posible de un modelo.
PrecauciónTarea
Queda como tarea del alumno modificar los hiperparámetros del modelo, buscando una mejora en su rendimiento
7.7 Naive Bayes
El clasificador ingenuo de Bayes (en inglés “Naïve Bayes”) es un algoritmo de clasificación basado en el conocido teorema de Bayes. A pesar de su simplicidad, los clasificadores “ingenuos” de Bayes son especialmente útiles para problemas con muchas variables de entrada, variables de entrada cuantitativas con un número muy grande de valores posibles y clasificación de texto. Es uno de los algoritmos básicos que suele aparecer en problemas de Procesamiento de Lenguaje Natural. Podrás estudiar aspectos fundamentales de Lenguaje Natural a lo largo de tus estudios de grado.
El teorema de Bayes relaciona la probabilidad condicional de dos eventos \(A\) y \(B\). La idea es modificar nuestras creencias iniciales sobre lo que ha sucedido (a priori) proporcionalmente con la forma en que nuestras observaciones se relacionan con sus posibles causas (probabilidad inversa):\[P(A∩B)=P(A,B)=P(A)*P(B|A)=P(B)*P(A|B)⇒P(B|A)=\frac{P(B)P(A|B)}{P(A)}\]
Su aplicación a un problema de clasificación es como sigue. Supongamos que queremos clasificar un conjunto de observaciones según una variable objetivo \(Y = \{y_1,y_2,\dots,y_K\}\), utilizando un vector de variables explicativas \(\mathbf{X} = (X_1,X_2,\ldots,X_p)\). La idea del modelo Naïve Bayes es estimar: \[P(Y=y_k|X_1=x_1,\dots,X_p=x_p)\] Mediante el teorema de Bayes: \[P(Y_k|\mathbf{X})=\frac{P(Y_k)P(X_1=x_1,\dots,X_p=x_p|Y_k)}{P(X_1=x_1,\dots,X_p=x_p)}\]
El paso clave del modelo Naïve Bayes es suponer que las variables explicativas son condicionalmente independientes dado el valor de la clase:\[P(X_1|X_2,\ldots,X_p,Y_k)=P(X_1|Y_k); P(X_2|X_3,\ldots,X_p,Y_k)=P(X_2|Y_k)\], etc. De forma general: \[P(X_1,X_2,\dots,X_p|Y_k)=\prod_{j=1}^{p} P(X_j | Y_k)\]
De este modo, aplicando el teorema de Bayes de manera recursiva, se tiene:
Esta es la expresión de la probabilidad de un valor de la variable respuesta dado el conjunto de valores de las variables explicativas. El denominador \(P(X_1,X_2,\ldots,X_p)\) es constante, de modo que bastará con considerar sólo el numerador para comparar las probabilidades de las diferentes clases condicionadas a los valores de las variables explicativas. Finalmente, la clase predicha es la que maximiza esta expresión: \[\arg \max_{k}\left\{ P(Y_k)\prod_{j=1}^{p} P(X_j | Y_k) \right\}\]
La probabilidad de clase \(P(Y_k)\) se denomina probabilidad a priori y se estima como la proporción de observaciones de la clase \(k\) en el conjunto de entrenamiento. Las probabilidades \(P(X_j|Y_k)\) son la verosimilitud. La verosimilitud mide cómo de verosímiles (creíbles) son las observaciones de las variables explicativas dado que se ha observado una determinada etiqueta en la variable respuesta. Las verosimilitudes se estiman en función del tipo de variable:
Si la variable \(X_j\) es cualitativa, para estimar la verosimilitud de la clase \(k\) con el predictor \(j\), se calcula la proporción de observaciones de la muestra de entrenamiento de la clase \(k\) en cada categoría de dicha variable.
Si la variable \(X_j\) es cuantitativa, habitualmente se asume una distribución Normal (también pueden utilizarse estimadores no paramétricos como histogramas) condicional a cada clase \(Y_k\), y se estiman los parámetros \(\mu_{jk}\), \(\sigma_{jk}\) para cada variable y clase: \[P(X_j=x_j|Y_k)=\frac{1}{\sqrt{2 \pi}\sigma_{jk}}\exp \left( - \frac{(x_j - \mu_{jk})^{2}}{2\sigma^{2}_{jk}} \right)\]
Finalmente se predice para cada conjunto de variables explicativas la clase con mayor probabilidad de clase condicionada \(P(Y_k|X_1,\ldots,X_p)\).
NotaIndependencia condicional
Una de las ideas clave del clasificador Naïve Bayes es que las variables explicativas \(X_1,\dots,X_p\) se asumen independientes entre sí dado el valor de la clase \(Y\). Formalmente: \[P(X_1,\dots,X_p | Y)= \prod_{j=1}^{p}P(X_j | Y)\]
Esto significa que, una vez conocemos la clase a la que pertenece una observación, el valor de una variable no aporta información adicional sobre las otras. Esta suposición simplifica enormemente el cálculo de las probabilidades conjuntas y hace que el modelo sea escalable incluso con muchas variables. A pesar de ser una suposición fuerte, en muchos contextos (como la clasificación de texto) funciona muy bien.
Ejemplo de independencia condicional Supongamos un escenario donde:
\(A\): Altura de una persona (por ejemplo, medida en centímetros).
\(V\): Vocabulario de una persona (por ejemplo, número de palabras conocidas).
\(E\): Edad de la persona (en años).
En la población general, la altura y el vocabulario no son independientes. Esto se debe a que las personas adultas suelen ser más altas y tener un vocabulario más amplio que los niños. Es decir, \(P(A, V) \neq P(A)P(V)\).
Sin embargo, si conocemos la edad de una persona (\(E\)), entonces la altura y el vocabulario son aproximadamente independientes dentro de cada grupo de edad. Por ejemplo:
Entre personas de 5 años, conocer la altura no da información adicional sobre el vocabulario.
Entre personas adultas, ocurre lo mismo.
Por tanto, podemos decir que existe entre \(A\) y \(V\) dado \(E\): \(A \perp V \mid E\)
Así, una vez conocida la edad, saber la altura no aporta información adicional sobre el vocabulario, y viceversa.
Formalmente, dadas tres variables aleatorias \(X\), \(Y\), y \(Z\), decimos que \(X\) es condicionalmente independiente de \(Y\) dado \(Z\), denotado como \(X \perp Y \mid Z\) si y solo si:
\[P(X \mid Y, Z) = P(X \mid Z)\]
para todo valor de \(Z\). En otras palabras, dado \(Z\), la distribución condicional de \(X\) no depende de \(Y\). Es decir, el conocimiento de \(Y\) no aporta información adicional sobre \(X\) una vez que sabemos \(Z\).
De manera equivalente, la independencia condicional puede expresarse como:
Lo que significa que la probabilidad conjunta de \(X\) y \(Y\), dada \(Z\), es el producto de las probabilidades condicionales de \(X\) y \(Y\) dadas \(Z\).
library(naivebayes)
naivebayes 1.0.0 loaded
For more information please visit:
https://majkamichal.github.io/naivebayes/
library(dplyr)library(caret)# Modelo Naive Bayesmodel <-naive_bayes(as.factor(deposit) ~ ., data = df.train, usekernel =TRUE)# Probabilidadesprobabilities <-predict(model, df.test %>% dplyr::select(-deposit), type ="prob")[,2]# Clasificaciónclasses <-as.numeric(probabilities >0.5)# Matriz de confusiónconfusionMatrix(table(classes, df.test$deposit),positive ="1")
Confusion Matrix and Statistics
classes 0 1
0 1156 266
1 312 1056
Accuracy : 0.7928
95% CI : (0.7773, 0.8077)
No Information Rate : 0.5262
P-Value [Acc > NIR] : < 2e-16
Kappa : 0.5852
Mcnemar's Test P-Value : 0.06124
Sensitivity : 0.7988
Specificity : 0.7875
Pos Pred Value : 0.7719
Neg Pred Value : 0.8129
Prevalence : 0.4738
Detection Rate : 0.3785
Detection Prevalence : 0.4903
Balanced Accuracy : 0.7931
'Positive' Class : 1
7.8 Modelos de mezcla de Gaussianas
Los modelos de mezcla de gaussianas (GMM, por sus siglas en inglés, “Gaussian Mixture Models”) son una técnica de ML utilizada para modelar datos con estructuras complejas y distribuciones múltiples. Estos modelos se basan en la idea de que un conjunto de datos puede estar compuesto por varias distribuciones gaussianas (también conocidas como normales). En otras palabras, los GMM permiten descomponer un conjunto de datos en múltiples componentes gaussianas, cada una de las cuales describe una parte de la distribución de los datos.
Los datos en el mundo real rara vez siguen una distribución simple y unimodal (¡ninguna teoría es tan compleja como un conjunto de datos!). Los GMM abordan esta complejidad al permitir que múltiples componentes gaussianas se superpongan y se combinen para representar mejor la verdadera distribución de los datos. Esto los convierte en una herramienta efectiva para modelar datos que pueden ser una mezcla de diferentes poblaciones o grupos subyacentes.
Cada componente gaussiana representa una subdistribución de datos, y se caracteriza por su media (promedio) y desviación estándar (dispersión). En un modelo de mezcla de gaussianas, se asume que cada componente es una distribución normal.
Cada observación en el conjunto se asigna a una de las componentes gaussianas en función de la probabilidad de que pertenezca a esa componente. Esto permite una descripción detallada de cómo se distribuyen los datos entre los diferentes grupos subyacentes.
Los GMM se utilizan en una variedad de aplicaciones, como la segmentación de imágenes, la detección de anomalías, la compresión de datos, la clasificación de patrones y la generación de datos sintéticos.
La estimación de los parámetros de un GMM, es decir, las medias y desviaciones estándar de las componentes gaussianas, se realiza mediante algoritmos de optimización que buscan maximizar la probabilidad conjunta de los datos observados.
Los GMM son especialmente útiles en la detección de anomalías, ya que permiten identificar regiones en el espacio de características donde los datos son inusuales o atípicos en comparación con el modelo GMM.
Aunque los GMM son efectivos en la modelación de datos complejos, pueden ser sensibles al número de componentes elegido y a la inicialización. La selección de un número adecuado de componentes y una buena inicialización son aspectos importantes en su aplicación.
8 Comparación de modelos
Hemos aplicado una variedad de modelos (y en otras asignaturas verás nuevos modelos de ML). Podemos comparar el rendimiento de modelos de diversas maneras. Una de ellas, tal y como vimos en el Capítulo 6 es la curva ROC:
library(pROC)roc_score
Call:
roc.default(response = bank.test$deposit, predictor = predicciones, plot = TRUE, print.auc = TRUE)
Data: predicciones in 1468 controls (bank.test$deposit no) < 1322 cases (bank.test$deposit yes).
Area under the curve: 0.6411
A la vista de los resultados. ¿Qué modelo es el más adecuado? Piensa sobre ello.
AdvertenciaModelo óptimo vs modelo útil
En este documento hemos aplicado una serie de modelos a un conjunto de datos. Nuestro objetivo es mostrar a los estudiantes varios métodos de ML. Nuestro objetivo nunca ha sido encontrar el modelo óptimo. Es decir, no buscamos el mejor modelo, sino un modelo útil. Un modelo que proporcione información y de ella podamos extraer conocimiento.
Agresti, Alan. 2015. Foundations of linear and generalized linear models. John Wiley & Sons.
Hastie, Trevor, Robert Tibshirani, Jerome H Friedman, y Jerome H Friedman. 2009. The elements of statistical learning: data mining, inference, and prediction. Vol. 2. Springer.
Kelleher, John D, y Brendan Tierney. 2018. Data science. MIT Press.
McCullagh, Peter. 2019. Generalized linear models. Routledge.
Murphy, Kevin P. 2012. Machine learning: a probabilistic perspective. MIT press.